
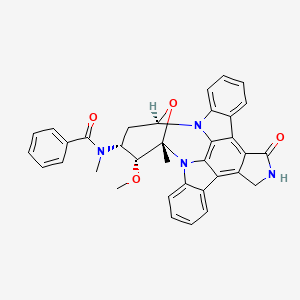
MIDOSTAURIN
(9S,10R,11R,13R)-2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3',2',1'-lm]pyrrolo[3,4-j][1,7]benzodiamzonine-1-one
N-[(9S,10R,11R,13R)-2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methylbenzamide
N-((9S,10R,11R,13R)-2,3,9,10,11,12-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo(1,2,3-gh:3',2',1'-lm)pyrrolo(3,4-j)(1,7)benzodiazonin-11-yl)-N-methyl-,
N-[(2R,4R,5R,6S)-5-methoxy-6-methyl-18-oxo-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-4-yl]-N-methylbenzamide hydrate
N-benzoyl staurosporine
NOVARTIS ONCOLOGY ORIGINATOR
Chemical Formula: C35H30N4O4
Exact Mass: 570.22671
Molecular Weight: 570.63710
Elemental Analysis: C, 73.67; H, 5.30; N, 9.82; O, 11.22
Tyrosine kinase inhibitors
PKC 412。PKC412A。CGP 41251。Benzoylstaurosporine;4'-N-Benzoylstaurosporine;Cgp 41251;Cgp 41 251.
120685-11-2 CAS
PHASE 3
- 4'-N-Benzoylstaurosporine
- Benzoylstaurosporine
- Cgp 41 251
- CGP 41251
- CGP-41251
- Midostaurin
- PKC 412
- PKC412
- UNII-ID912S5VON
Midostaurin is an inhibitor of tyrosine kinase, protein kinase C, and VEGF. Midostaurin inhibits cell growth and phosphorylation of FLT3, STAT5, and ERK. It is a potent inhibitor of a spectrum of FLT3 activation loop mutations.

it is prepared by acylation of the alkaloid staurosporine (I) with benzoyl chloride (II) in the presence of diisopropylethylamine in chloroform.
Midostaurin is a synthetic indolocarbazole multikinase inhibitor with potential antiangiogenic and antineoplastic activities. Midostaurin inhibits protein kinase C alpha (PKCalpha), vascular endothelial growth factor receptor 2 (VEGFR2), c-kit, platelet-derived growth factor receptor (PDGFR) and FMS-like tyrosine kinase 3 (FLT3) tyrosine kinases, which may result in disruption of the cell cycle, inhibition of proliferation, apoptosis, and inhibition of angiogenesis in susceptible tumors.
Derivative of staurosporin, orally active, potent inhibitor of FLT3 tyrosine kinase (fetal liver tyrosine kinase 3). In addition Midostaurin inhibits further molecular targets such as VEGFR-1 (Vascular Endothelial Growth Factor Receptor 1), c-kit (stem cell factor receptor), H-and K-RAS (Rat Sarcoma Viral homologue) and MDR (multidrug resistance protein).
Midostaurin inhibits both wild-type FLT3 and FLT3 mutant, wherein the internal tandem duplication mutations (FLT3-ITD), and the point mutation to be inhibited in the tyrosine kinase domain of the molecule at positions 835 and 836.Midostaurin is tested in patients with AML.

Midostaurin, a protein kinase C (PKC) and Flt3 (FLK2/STK1) inhibitor, is in phase III clinical development at originator Novartis for the oral treatment of acute myeloid leukemia (AML).
Novartis is conducting phase III clinical trials for the treatment of aggressive systemic mastocytosis or mast cell leukemia. The National Cancer Institute (NCI) is conducting phase I/II trials with the drug for the treatment of chronic myelomonocytic leukemia (CMML) and myelodysplastic syndrome (MDS).
Massachusetts General Hospital is conducting phase I clinical trials for the treatment of adenocarcinoma of the rectum in combination with radiation and standard chemotherapy.
 MIDOSTAURIN
MIDOSTAURIN
Midostaurin (PKC412) is a multi-target protein kinase inhibitor being investigated for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). It is a semi-synthetic derivative of staurosporine, an alkaloid from the bacterium Streptomyces staurosporeus, and is active in patients with mutations of CD135 (FMS-like tyrosine kinase 3 receptor).[1]
After successful Phase II clinical trials, a Phase III trial for AML has started in 2008. It is testing midostaurin in combination with daunorubicin and cytarabine.[2] In another trial, the substance has proven ineffective in metastatic melanoma.[3]
Midostaurin has also been studied at Johns Hopkins University for the treatment of age-related macular degeneration (AMD), but no recent progress reports for this indication have been made available. Trials in macular edema of diabetic origin were discontinued at Novartis.
In 2004, orphan drug designation was received in the E.U. for the treatment of AML. In 2009 and 2010, orphan drug designation was assigned for the treatment of acute myeloid leukemia and for the treatment of mastocytosis, respectively, in the U.S. In 2010, orphan drug designation was assigned in the E.U. for the latter indication.
 MIDOSTAURIN
MIDOSTAURINReferences
- Fischer, T.; Stone, R. M.; Deangelo, D. J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E. J.; Schiller, G. J.; Klimek, V. M.; Nimer, S. D.; Gilliland, D. G.; Dutreix, C.; Huntsman-Labed, A.; Virkus, J.; Giles, F. J. (2010). "Phase IIB Trial of Oral Midostaurin (PKC412), the FMS-Like Tyrosine Kinase 3 Receptor (FLT3) and Multi-Targeted Kinase Inhibitor, in Patients with Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome with Either Wild-Type or Mutated FLT3". Journal of Clinical Oncology 28 (28): 4339–4345. doi:10.1200/JCO.2010.28.9678. PMID 20733134. edit
- ClinicalTrials.gov NCT00651261 Daunorubicin, Cytarabine, and Midostaurin in Treating Patients With Newly Diagnosed Acute Myeloid Leukemia
- Millward, M. J.; House, C.; Bowtell, D.; Webster, L.; Olver, I. N.; Gore, M.; Copeman, M.; Lynch, K.; Yap, A.; Wang, Y.; Cohen, P. S.; Zalcberg, J. (2006). "The multikinase inhibitor midostaurin (PKC412A) lacks activity in metastatic melanoma: a phase IIA clinical and biologic study". British Journal of Cancer 95 (7): 829–834. doi:10.1038/sj.bjc.6603331. PMC 2360547. PMID 16969355.
- Midostaurin product page, Fermentek
- Wang, Y; Yin, OQ; Graf, P; Kisicki, JC; Schran, H (2008). "Dose- and Time-Dependent Pharmacokinetics of Midostaurin in Patients With Diabetes Mellitus". J Clin Pharmacol 48 (6): 763–775. doi:10.1177/0091270008318006. PMID 18508951.
- Ryan KS (2008). "Structural studies of rebeccamycin, staurosporine, and violacein biosynthetic enzymes". Ph.D. Thesis. Massachusetts Institute of Technology.
Bioorg Med Chem Lett 1994, 4(3): 399
US 5093330
EP 0657164
EP 0711556
EP 0733358
WO 1998007415
WO 2002076432
WO 2003024420
WO 2003037347
WO 2004112794
WO 2005027910
WO 2005040415
WO 2006024494
WO 2006048296
WO 2006061199
WO 2007017497
WO 2013086133
WO 2012016050
WO 2011000811
3-1-2010
|
Colony stimulating factor-1 receptor as a target for small molecule inhibitors.
|
Bioorganic & medicinal chemistry
|
7-18-2012
|
Staurosporine Derivatives as Inhibitors of FLT3 Receptor Tyrosine Kinase Activity
| |
6-13-2012
|
Crystal form of N-benzoyl-staurosporine
| |
12-14-2011
|
COMPOSITIONS FOR TREATMENT OF SYSTEMIC MASTOCYTOSIS
| |
7-6-2011
|
Staurosporine derivatives as inhibitors of flt3 receptor tyrosine kinase activity
| |
7-6-2011
|
Staurosporine Derivatives for Use in Alveolar Rhabdomyosarcoma
| |
12-10-2010
|
Pharmaceutical Compositions for treating wouds and related methods
| |
11-5-2010
|
COMBINATIONS OF JAK INHIBITORS
| |
7-23-2010
|
COMBINATIONS COMPRISING STAUROSPORINES
| |
3-5-2010
|
COMBINATION OF IAP INHIBITORS AND FLT3 INHIBITORS
| |
1-29-2010
|
ANTI-CANCER PHOSPHONATE ANALOGS
|
1-13-2010
|
Therapeutic phosphonate compounds
| |
11-20-2009
|
Use of Staurosporine Derivatives for the Treatment of Multiple Myeloma
| |
7-17-2009
|
KINASE INHIBITORY PHOSPHONATE ANALOGS
| |
6-19-2009
|
Organic Compounds
| |
3-20-2009
|
Use of Midostaurin for Treating Gastrointestinal Stromal Tumors
| |
11-21-2008
|
PHARMACEUTICAL COMPOSITIONS COMPRISING A POORLY WATER-SOLUBLE ACTIVE INGREDIENT, A SURFACTANT AND A WATER-SOLUBLE POLYMER
| |
11-19-2008
|
Anti-cancer phosphonate analogs
| |
9-12-2008
|
Multi-Functional Small Molecules as Anti-Proliferative Agents
| |
9-5-2008
|
Sensitization of Drug-Resistant Lung Caners to Protein Kinase Inhibitors
| |
8-29-2008
|
Organic Compounds
|
8-27-2008
|
Kinase inhibitory phosphonate analogs
| |
4-25-2008
|
Treatment Of Gastrointestinal Stromal Tumors With Imatinib And Midostaurin
| |
12-28-2007
|
Pharmaceutical Uses of Staurosporine Derivatives
| |
12-7-2007
|
Kinase Inhibitor Phosphonate Conjugates
| |
8-17-2007
|
Combinations comprising staurosporines
| |
10-13-2006
|
Staurosporine derivatives for hypereosinophilic syndrome
| |
7-15-2005
|
Phosphonate substituted kinase inhibitors
| |
10-20-2004
|
Staurosporin derivatives
|
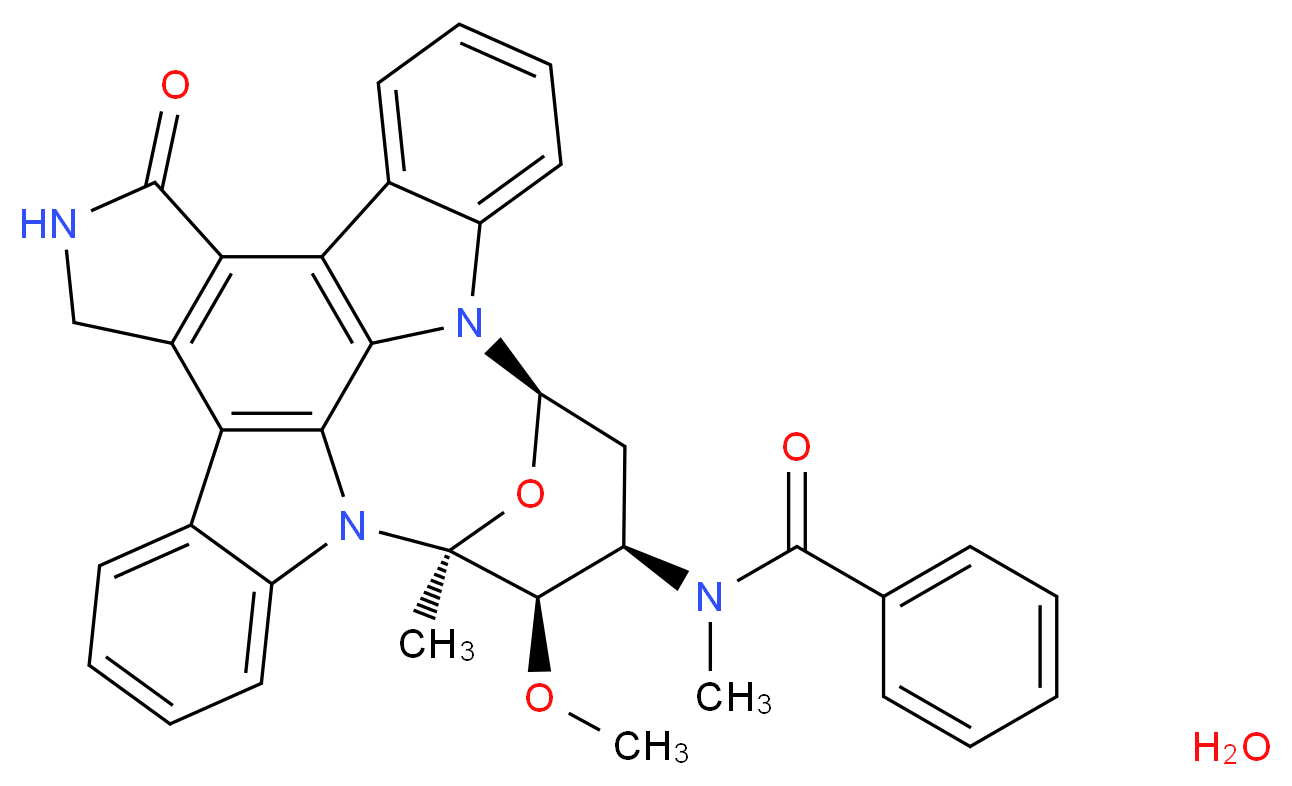
MIDOSTAURIN HYDRATE
Midostaurin according to the invention is N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methylbenzamide of the formula (II):

or a salt thereof, hereinafter: “Compound of formula II or midostaurin”.
Compound of formula II or midostaurin [International Nonproprietary Name] is also known as PKC412.
Midostaurin is a derivative of the naturally occurring alkaloid staurosporine, and has been specifically described in the European patent No. 0 296 110 published on Dec. 21, 1988, as well as in U.S. Pat. No. 5093330 published on Mar. 3, 1992, and Japanese Patent No. 2 708 047.
......................
The nomenclature of the products is, on the complete structure of staurosporine ([storage]-NH-CH ₃derived, and which is designated by N-substituent on the nitrogen of the methylamino group

Example 18:
- N-Benzoyl-staurospor
- A solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N, N-diisopropylethylamine in 2 ml of chloroform is added at room temperature with 0.035 ml (0.3 mmol) of benzoyl chloride and 10 stirred minutes.The reaction mixture is diluted with chloroform, washed with sodium bicarbonate, dried over magnesium sulfate and evaporated. The crude product is chromatographed on silica gel (eluent methylene chloride / ethanol 30:1), mp 235-247 ° with brown coloration.
- cut paste may not be ok below
Staurosporine the formula [storage]-NH-CH ₃ (II) (for the meaning of the rest of [storage] see above) as the basic material of the novel compounds was already in 1977, from the cultures of Streptomyces staurosporeus AWAYA, and TAKAHASHI

MURA, sp. nov. AM 2282, see Omura, S., Iwai, Y., Hirano, A., Nakagawa, A.; awayâ, J., Tsuchiya, H., Takahashi, Y., and Masuma, R. J. Antibiot. 30, 275-281 (1977) isolated and tested for antimicrobial activity. It was also found here that the compound against yeast-like fungi and microorganisms is effective (MIC of about 3-25 mcg / ml), taking as the hydrochloride = having a LD ₅ ₀ 6.6 mg / kg (mouse, intraperitoneal). Stagnated recently it has been shown in extensive screening, see Tamaoki, T., Nomoto, H., Takahashi, I., Kato, Y, Morimoto, M. and Tomita, F.: Biochem. and Biophys. Research Commun. 135 (No. 2), 397-402 (1986) that the compound exerts a potent inhibitory effect on protein kinase C (rat brain)
.....................
EXAMPLE 18 N-benzoyl-staurosporine
0.035 ml (0.3 mmol) of benzoyl chloride is added at room temperature to a solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N,N-diisopropylethylamine in 2 ml of chloroform and the whole is stirred for 10 minutes. The reaction mixture is diluted with chloroform, washed with sodium bicarbonate solution, dried over magnesium sulphate and concentrated by evaporation. The crude product is chromatographed on silica gel (eluant:methylene chloride/ethanol 30:1); m.p. 235
.........................
Bioorg Med Chem Lett 1994, 4(3): 399

........................
A variety of PKC inhibitors are available in the art for use in the invention. These include bryostatin (U.S. Patent 4,560,774), safinogel (WO 9617603), fasudil (EP 187371), 7- hydoxystaurosporin (EP 137632B), various diones described in EP 657458, EP 657411 and WO9535294, phenylmethyl hexanamides as described in WO9517888, various indane containing benzamides as described in WO9530640, various pyrrolo [3,4-c]carbazoles as described in EP 695755, LY 333531 (IMSworld R & D Focus 960722, July 22, 1996 and Pharmaprojects Accession No. 24174), SPC-104065 (Pharmaprojects Accession No. 22568), P-10050 (Pharmaprojects Accession No. 22643), No. 4432 (Pharmaprojects Accession No. 23031), No. 4503 (Pharmaprojects Accession No. 23252), No. 4721 (Pharmaprojects Accession No. 23890), No. 4755 (Pharmaprojects Accession No. 24035), balanol (Pharmaprojects Accession No. 20376), K-7259 (Pharmaprojects Accession No. 16649), Protein kinase C inhib, Lilly (Pharmaprojects Accession No. 18006), and UCN-01 (Pharmaprojects Accession No. 11915). Also see, for example, Tamaoki and Nakano (1990) Biotechnology 8:732-735; Posada et al. (1989) Cancer Commun. 1:285-292; Sato et al. (1990) Biochem Biophys. Res. Commun. 173:1252-1257; Utz et al. (1994) Int. J. Cancer 57:104-110; Schwartz et al. (1993) J. Na . Cancer lnst. 85:402-407; Meyer et al. (1989) Int. J. Cancer 43:851-856; Akinaga et al. (1991) Cancer Res. 51:4888-4892, which disclosures are herein incorporated by reference. Additionally, antisense molecules can be used as PKC inhibitors. Although such antisense molecules inhibit mRNA translation into the PKC protein, such antisense molecules are considered PKC inhibitors for purposes of this invention. Such antisense molecules against PKC inhibitors include those described in published PCT patent applications WO 93/19203, WO 95/03833 and WO 95/02069, herein incorporated by reference. Such inhibitors can be used in formulations for local delivery to prevent cellular proliferation. Such inhibitors find particular use in local delivery for preventing rumor growth and restenosis.
N-benzoyl staurosporine is a benzoyl derivative of the naturally occurring alkaloid staurosporine. It is chiral compound ([a]D=+148.0+-2.0°) with the formula C35H30R1O4 (molecular weight 570.65). It is a pale yellow amorphous powder which remains unchanged up to 220°C. The compound is very lipophilic (log P>5.48) and almost insoluble in water (0.068 mg/1) but dissolves readily in DMSO.
............................
staurosporine
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus. It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive. The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment

Staurosporine is the precursor of the novel protein kinase inhibitor midostaurin(PKC412). Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
Indolocarbazoles belong to the alkaloid sub-class of bisindoles. Of these carbazoles the Indolo(2,3-a)carbazoles are the most frequently isolated; the most common subgroup of the Indolo(2,3-a)carbazoles are the Indolo(2,3-a)pyrrole(3,4-c)carbazoles which can be divided into two major classes - halogenated (chlorinated) with a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond and the second class consists of both indole nitrogen glycosilated, non-halogenated, and a fully reduced C-7 carbon. Staurosporine is part of the second non-halogenated class.
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imineby enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450enzyme to form an aromatic ring system occurs

This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4'amine by StaMA and N-methylation of the 3'-hydroxy by StaMB leads to the formation of staurosporine
| US4107297 * | 28 Nov 1977 | 15 Aug 1978 | The Kitasato Institute | Antibiotic compound |
| US4735939 * | 27 Feb 1987 | 5 Apr 1988 | The Dow Chemical Company | Insecticidal activity of staurosporine |
| ZA884238A * | Title not available |
No comments:
Post a Comment