GRAVIR SERIES

1 RALTEGRAVIR
2 ELVITEGRAVIR
3.DOLUTEGRAVIR
4 GS 9883, Bictegravir
5
6
7
1. RALTEGRAVIR










 Corresponding author email
Corresponding author email



 -2012 August anti-AIDS drugs approved by the FDA")

.jpg)


















1 RALTEGRAVIR
2 ELVITEGRAVIR
3.DOLUTEGRAVIR
4 GS 9883, Bictegravir
5
6
7
1. RALTEGRAVIR
CAS No…….518048-05-0 (free acid)
871038-72-1 (monopotassium salt)IUPAC Name:- N-(2-(4-(4-fluorobenzylcarbamoyl)-5-hydroxy-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)propan-2-yl)
871038-72-1 (monopotassium salt)IUPAC Name:- N-(2-(4-(4-fluorobenzylcarbamoyl)-5-hydroxy-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)propan-2-yl)
Organic Process Research and Development, 2011 , vol. 15, 1 pg. 73 – 83,
143 – 144.1 °C(free acid)
MW: 444.42
………………………………………………….
K SALT
C20H20FN6O5*K, 482.513
MP..275 – 277 °C
European Journal of Medicinal Chemistry, 2012 , vol. 50, pG. 361 – 369
Drug information:- Raltegravir is an Anti-microbial drug further classified as anti-viral agent of the class integrase inhibitor. It is used either signally or in combination with other drugs for the treatment of human immunodeficiency virus (HIV) and further clinical trials are in process.
Raltegravir (RAL, Isentress, formerly MK-0518) is an antiretroviral drug produced by Merck & Co., used to treat HIV infection.[1] It received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval.[2][3]
In December 2011, it received FDA approval for pediatric use in patients ages 2–18, taken in pill form orally twice a day by prescription with two other antiretroviral medications to form the cocktail (most anti-HIV drugs regimens for adults and children use these cocktails). Raltegravir is available in chewable form but- because the two tablet formulations are not interchangeable- the chewable pills are only approved for use in children two to 11. Older adolescents will use the adult formulation.[4]
Raltegravir targets integrase, an HIV enzyme that integrates the viral genetic material into human chromosomes, a critical step in the pathogenesis of HIV. The drug is metabolized away via glucuronidation.[5]

Isentress tablets
Raltegravir is taken orally twice daily.[3] Doses of 200, 400, and 600 mg have been studied.
At the 2007 Conference on Retroviruses and Opportunistic Infections, researchers presented Phase III data showing that 77% of patients taking the 400 mg dose of raltegravir plus other antiretroviral drugs reached HIV viral loads below 400 copies, nearly twice as many compared with a control group.
Raltegravir was initially approved only for use in individuals whose infection has proven resistant to otherHAART drugs.[3] However, in July 2009, the FDA granted expanded approval for Raltegravir for use in all patients.[6] As with any HAART medication, raltegravir is unlikely to show durability if used as monotherapy.
In a study of the drug as part of combination therapy, raltegravir exhibited potent and durable antiretroviral activity similar to that of efavirenz at 24 and 48 weeks but achieved HIV-1 RNA levels below detection at a more rapid rate. After 24 and 48 weeks of treatment, raltegravir did not result in increased serum levels of total cholesterol, low-density lipoprotein cholesterol, or triglycerides.[7][8]
Raltegravir significantly alters HIV viral dynamics and decay and further research in this area is ongoing. In clinical trials patients taking raltegravir achieved viral loads less than 50 copies per millitre sooner than those taking similarly potent Non-nucleoside Reverse Transcriptase Inhibitors orProtease Inhibitors. This statistically significant difference in viral load reduction has caused some HIV researchers to begin questioning long held paradigms about HIV viral dynamics and decay.[9] Research into raltegravir’s ability to affect latent viral reservoirs and possibly aid in the eradication of HIV is currently ongoing.[10]
Research results were published in the New England Journal of Medicine on July 24, 2008. The authors concluded that “raltegravir plus optimized background therapy provided better viral suppression than optimized background therapy alone for at least 48 weeks.” [11]
Research on human cytomegalovirus (HCMV) terminase proteins demonstrated that Raltegravir may block viral replication of the herpesviruses.[12]
In January 2013, a Phase II trial was initiated to evaluate the therapeutic benefit of raltegravir in treating multiple sclerosis (MS).[13] The drug is active against Human Endogenous Retroviruses(HERVs) and possibly Epstein-Barr Virus, which have been suggested in the pathogenesis of relapsing-remitting MS.
Raltegravir was generally well tolerated when used in combination with optimized background therapy regimens in treatment-experienced patients with HIV-1 infection in trials of up to 48 weeks’ duration.[14]
Synthesis


WO 2006060730
…………………………………………………
- Savarino A (December 2006). “A historical sketch of the discovery and development of HIV-1 integrase inhibitors”. Expert Opin Investig Drugs 15 (12): 1507–22. doi:10.1517/13543784.15.12.1507.PMID 17107277.
- “FDA approval of Isentress (raltegravir)”. U.S. Food and Drug Administration (FDA). June 25, 2009. Retrieved 2009-11-15.
- “Isentress Drug Approval Package”. U.S. Food and Drug Administration (FDA). February 22, 2008. Retrieved 2009-11-15.
- http://www.everydayhealth.com/hiv-aids/1222/fda-okays-raltegravir-for-kids-teens-with-hiv.aspx?xid=aol_eh-hiv_6_20111219_&aolcat=HLT&icid=maing-grid7%7Cmain5%7Cdl10%7Csec3_lnk2%26pLid%3D122480
- HIV Antiretroviral Agents in Development
- “UPDATE 2-FDA OKs widened use of Merck’s Isentress HIV drug”. Reuters. 2009-07-10.
- Markowitz M, Nguyen BY, Gotuzzo E, et al. (2007). “Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study”. J. Acquir. Immune Defic. Syndr. 46 (2): 125–33. doi:10.1097/QAI.0b013e318157131c. PMID 17721395.
- Stephenson J (2007). “Researchers buoyed by novel HIV drugs: will expand drug arsenal against resistant virus”. JAMA 297 (14): 1535–6. doi:10.1001/jama.297.14.1535. PMID 17426263.
- Faster Viral Decay With Raltegravir
- ClinicalTrials.gov NCT00554398 Impact of MK-0518 (Raltegravir) Intensification on HIV-1 Viral Latency in Patients With Previous Complete Viral Suppression
- Steigbigel RT, Cooper DA, Kumar PN, et al. (July 2008). “Raltegravir with optimized background therapy for resistant HIV-1 infection”. N. Engl. J. Med. 359 (4): 339–54.doi:10.1056/NEJMoa0708975. PMID 18650512.
- Drug against AIDS could be effective against herpesvirus
- Raltegravir (Isentress) Pilot Study in Relapsing Multiple Sclerosis (INSPIRE)
- Croxtall JD, Keam SJ. (2009). “Raltegravir”. Drugs 69 (8): 1059–75. doi:10.2165/00003495-200969080-00007. PMID 19496631.
- Belyk, K. M.; Morrison, H. G.; Jones, P.; Summa, V.; 2007, WO 2006060730
- Manufacturer’s website
- MK-0518 at Aidsmedscom[dead link]
- Integrase Inhibitor Raltegravir (MK-0518) Doubles HIV Suppression in Treatment-Experienced Patients (aidsmap 28 February 2007)
- RMK-0518 Abstract from CROI 2007
- Interim Results From Phase II Study Of MK-0518
- World patent covering the potassium salt
- Raltegravir Pharmacokinetics
……………………………………………………………………………………………………………………………………………….
Raltegravir, also referred to as Raltegravir free-hydroxy, N-(2-(4-(4-fluorobenzyl- carbamoyl)-5-hydroxy-l-methyl-6-oxo-l ,6-dihydropyrimidin-2-yl)propan-2-yl)-5-methyl- l ,3,4-oxadiazole-2-carboxamide, having the following structure;
is an antiretroviral drug used to treat HIV infection. Raltegravir targets integrase, an HIV enzyme that integrates the viral genetic material into human chromosomes, a critical step in the pathogenesis of HIV. Raltegravir potassium salt is marketed under the trade name ISENTRESS™ by Merck & Co.
The processes for preparing Raltegravir that are known in the art either require a protection step for the 5-hydroxy group prior to the methylation step, or lead to an impurity resulting from the methylation of the 5-hydroxy group.
U.S. Patent No. 7, 169,780 discloses Raltegravir and preparation thereof, as described in the following reaction scheme:
Scheme 1
J. Med. Chem. 2008, 51 , 5843-5855 discloses another process for preparing Raltegravir as described in the following reaction scheme:
RLT K-salt
Scheme 2 U.S. Publication No. US 2006/0122205 describes an alternative process for preparing Raltegravir, in which the alkylation step does not include a step for protecting the 5-hydroxy group. The process is described in the following reaction scheme:
Scheme 3
Provided herein is an industrially applicable process for preparing RLT-7′, RLT-8, RLT-9 and RLT-9-OP, intermediates in the synthesis of Raltegravir, as well as processes for preparing Raltegravir and crystalline forms thereof.
US Publication No. US 2006/0122205, WO 2010/1401 56 and WO 201 1 /
024192 describe the potassium salt of Raltegravir, including amorphous and crystalline forms I, II, III and H I , as well as amorphous and crystalline forms of Raltegravir free- hydroxy. PCT publication No. WO 201 1/123754 describes certain Raltegravir salts and polymorphs, including form V of Raltegravir potassium.
Conditions:-
i. Benzylchloroformate, N,N-diisopropylethylamine, Methyl tert-butyl ether, 20 – 25 °C, 16 h, ii. Hydroxyl amine, Water, 60 °C, 3 h, iii. Dimethyl acetylenedicarboxylate, methanol, Room temperature 2 h then Xylene 90 °C, 2 h, iv. Magnesium methoxide, dimethyl sulfoxide, Methyl iodide, 20 – 25 °C, 2 h, v. 4-fluorobenzyl amine, ethanol, 72 °C, 2 h, vi. 5% Pd/C, methanol, Molybdate sulfuric acid, Hydrogen gas, 50 °C, 3 h, vii. 5-methyl-1,3,4-oxadiazole-2-carbonylchloride, N-methylmorpholine, Tetrahydrofuran, 0 – 5 °C, 2 h
|
preparation of Raltegravir is described in US patent 2006122205A1 and also in WO2006060730. Accordingly, 2-amino-2-methyl-propanenitrile 1 was reacted with benzylchloroformate in presence of N,N-diisopropylethylamine using methyl tert-butyl ether as solvent at ambient temperature to give benzyl N-(1-cyano-1-methyl-ethyl)carbamate 2. Treatment of 2 with hydroxyl amine using water as solvent at elevated temperature give benzyl N-[(2Z)-2-amino-2-hydroxyimino-1,1-dimethyl-ethyl]carbamate 3. The compound 3 was further cyclized with dimethyl acetylenedicarboxylate using methanol as solvent at higher temperature to give methyl 2-(1-benzyloxycarbonylamino-1-methyl-ethyl)-5-hydroxy-6-oxo-1H-pyrimidine-4-carboxylate 4. Compound 4 was then methylated with methyl iodide in presence of magnesium methoxide as base and dimethyl sulfoxide as solvent at ambient temperature to give methyl 2-(1-benzyloxycarbonylamino-1-methyl-ethyl)-5-hydroxy-1-methyl-6-oxo-pyrimidine-4-carboxylate 5. Compound 5 on condensing with 4-fluorobenzyl amine using ethanol as solvent result in to benzyl N-[1-[4-[(4-fluorophenyl)methylcarbamoyl]-5-hydroxy-1-methyl-6-oxo-pyrimidin-2-yl]-1-methyl-ethyl]carbamate 6, which underwent benzyloxy-decarboxylation on hydrogenating with hydrogen gas in presence of 5% Palladium on carbon catalyst and molybdate sulfuric acid using methanol as solvent to give 2-(1-amino-1-methyl-ethyl)-N-[(4-fluorophenyl)methyl]-5-hydroxy-1-methyl-6-oxo-pyrimidine-4-carboxamide 7. The final step involves condensation of 7 with 5-methyl-1,3,4-oxadiazole-2-carbonylchloride in presence of N-methylmorpholine as base using tetrahydrofuran as solvent at slightly lower temperature to afford N-[1-[4-[(4-fluorophenyl)methylcarbamoyl]-5-hydroxy-1-methyl-6-oxo-pyrimidin-2-yl]-1-methyl-ethyl]-5-methyl-1,3,4-oxadiazole-2-carboxamide also called Raltegravir 8.
|
The formation of the hydroxypyrimidone core (3.22) of raltegravir deserves further discussion as its unexpected mechanism was only recently fully elucidated in a joint effort between Merck process chemists and the Houk group at UCLA [91]. These studies combined B3LYP density functional theory with labelling studies and revealed that the most likely pathway involves the formation of a tightly bound polar radical pair 3.31 resulting from thermal homolysis of the N–O bond (Scheme 35). This species subsequently recombines under formation of a C–N bond and a C=O double bond (3.32) allowing for the final cyclocondensation to occur with liberation of methanol. Furthermore these studies were able to disprove a potential alternative [3,3]-sigmatropic rearrangement step by incorporating 15N enriched precursors leading to the formation of pyrimidone 3.22, which is only consistent with a formal [1,3]-sigmatropic rearrangement. Subsequent calculations demonstrated the high energy barrier for such a concerted [1,3]-shift, ultimately leading to the finding of the before-mentioned polar radical pair pathway which is about 8 kcal/mol lower in energy. This is consistent with the experimentally observed rate acceleration in case of the Z-isomer of 3.33 over the E-isomer which was also confirmed by calculations showing an energy gap of 3 kcal/mol.
An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles
Marcus Baumann and Ian R. Baxendale
and Ian R. Baxendale
Department of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK
Corresponding author email
Associate Editor: P. R. Hanson
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
check beilstein journals as per link above ……………
this publication allows free usage of data if given proper ref…………………..
any objections email me amcrasto@gmail.com or cal +91 9323115463
………………..
nmr
Imp roved synthesis of raltegravir
GUO D i2liang et al
Department ofM edicinal Chem istry, China PharmaceuticalUniversity, N anjing 210009;
Journal of China Pharmaceutical University 2009, 40 (4) : 297 – 301
1H NMR (CD3OD) δ: 7.40 (m, 2H) , 7.04 (m , 2H) ,
4.56 (s, 2H ) , 3.46 ( s, 3H ) , 2.65 (s, 3H ) , 1.83 (s,
6H);
13C NMR (CD3OD ) δ: 168.4, 164.8, 163.2,
162.0, 161.9, 160.1, 155.3, 145.8, 136.0, 134.9,
131.0, 116.7, 116.6, 60.2, 43.8, 41.3, 34.8, 27.6,
11.4;
ESI2MS m /z 443 (M )-; LR2MS (EI) m /z 444(M )+; HR2MS ( E I) m /z C20 H21 FN6O5(M )+
calcd444, 155, 7, found 444, 154, 2
second set
lH NMR (399.87 MHz5 CDCI3) δ 12.04 (s, IH), 8.45 (s, IH), 7.94 (t, J = 6.2 Hz, IH), 7.41-736 (m, 2H), 7.08-7.02 (m, 2H)5 4.61 (d, J – 6.2 Hz, 2H), 3.68 (s, 3H), 2.63 (s, 3H), 1.87 (s, 6H).
13C NMR (100.55 MHz, CDCI3) δ 168.3, 166.7, 162.6 (d, JCF=245.7 Hz), 159.6, 159.1, 152.O5 150.4, 147.2, 133.4 (d, JCP=3.2 Hz)5129.9 (d, JcF=8.0 Hz), 124.1, 115.9 (d, JcF=21.7 Hz), 58.0, 42.7, 33.5, 26.7, 11.4.
…………………….
IR
absorption bandsKBR (cm“1) at 832, 1017, 1248, 1350, 1510, 1682, 2995, and 3374
……………….
K SALT
Org. Process Res. Dev., 2011, 15 (1), pp 73–83
DOI: 10.1021/op100257r
mp 274.2−275.2 °C. 1H NMR (500 MHz, DMSO-d6) δ: 11.65 (t, J = 6.0 Hz, 1 H), 9.75 (s, 1 H), 7.36 (dd, J = 8.6, 5.7 Hz, 2 H), 7.14 (app. t,J = 8.6 Hz, 2 H), 4.48 (d, J = 6.0 Hz, 2 H), 3.43 (s, 3 H), 2.58 (s, 3 H), 1.73 (s, 6 H);
13C NMR (125 MHz, DMSO-d6) δ: 168.7, 167.0, 166.6, 162.1 (d, JCF = 243 Hz), 159.7, 158.3, 153.1, 139.6, 138.0 (d, JCF = 3 Hz), 130.2 (d, JCF = 8 Hz), 123.7, 116.0 (d, JCF = 22), 58.4, 42.1, 33.3, 28.1 (2 C), 11.7.
……………………………
impurities
Org. Process Res. Dev., 2012, 16 (8), pp 1422–1429
DOI: 10.1021/op300077m
…………………
intermediates
N-[(1Z)-1 -amino-1 -(hydroxyimino)-2-memylpropan-2-yl]-5-methyl-l ,3 ,4- oxadiazole-2-carboxamide (IVa) (198 gms) was suspended in methanol (1188 ml) and cooled to 15 to 25°C. Dimethyl acetyl enedicarboxylate (DMAD; 152.8 gms) was added and the reaction mass was stirred for 2 to 3 hours at 25°C. The reaction mass was concentrated under reduced pressure and xylene was added and stirred between 135°C and 125°C for 6 hour. After completion of reaction, the mixture was cooled to 60°C and methanol (170 ml) & methyl tert-butyl ether (MTBE) were added to the reaction mass and stirred for 1 hour. The resultant slurry was filtered and washed with a 9:1 mixture of methanol & methyl tert-butyl ether (MTBE) and dried to give methyl 2-(2-(5-methyl-l ,3,4-oxadiazole-2-carboxamido)propan-2-yl)-l ,6-dihydro-5- hydroxy-6-oxopyrimidine-4-carboxylate (V a).
Yield: 198 gms (66 %).
1H NMR (400 MHz, DMSO d6): δ 12.74 (s, 1H), 10.35 (s, 1H), 9.12 (s, 1 H), 3.81 (s, 3H), 2.58 (s, 3H), 1.59 (s, 6 H);
13C NMR (100 MHz, DMSO d6): δ 166.60, 166.15, 160.19, 159.23, 153.26, 152.87, 145.65, 128.30, 56.60, 52.91 , 26.26, 11.34;
retroviral drugs
elvitegravir
Å for chemical synthesis from carboxylic acids elvitegravir 1 starts, the NIS transformed into acid chloride iodide, 2 , and with 3 condensation 4 . 4 and amino alcohols 5 addition-elimination reaction occurs 6 , 6 in alkaline conditions Shimonoseki ring hydroxyl group protected with TBS after seven , seven and zinc reagent 8 occurred Negishi coupling get nine , the last ninehydrolysis and methoxylated get angstrom for elvitegravir.
.................................
2. ELVITEGRAVIR
Elvitegravir
697761-98-1 CAS
FOSTER CITY, Calif.–(BUSINESS WIRE)–Nov. 18, 2013– Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission has granted marketing authorization for VitektaTM (elvitegravir 85 mg and 150 mg) tablets, an integrase inhibitor for the treatment of HIV-1 infection in adults without known mutations associated with resistance to elvitegravir. Vitekta is indicated for use as part of HIV treatment regimens that include a ritonavir-boosted protease inhibitor.http://www.pharmalive.com/eu-oks-gileads-vitektaVitekta interferes with HIV replication by blocking the virus from integrating into the genetic material of human cells. In clinical trials, Vitekta was effective in suppressing HIV among patients with drug-resistant strains of HIV.http://www.pharmalive.com/eu-oks-gileads-vitekta
Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).

- Gilead Press Release Phase III Clinical Trial of Elvitegravir July 22, 2008
- Gilead Press Release Gilead and Japan Tobacco Sign Licensing Agreement for Novel HIV Integrase Inhibitor March 22, 2008
- Shimura K, Kodama E, Sakagami Y, et al. (2007). “Broad Anti-Retroviral Activity and Resistance Profile of a Novel Human Immunodeficiency Virus Integrase Inhibitor, Elvitegravir (JTK-303/GS-9137)”. J Virol 82 (2): 764. doi:10.1128/JVI.01534-07. PMC 2224569. PMID 17977962.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Sax, P. E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J. E.; Liu, H. C.; Zhong, L.; Yale, K.; White, K.; Kearney, B. P.; Szwarcberg, J.; Quirk, E.; Cheng, A. K.; Gs-Us-236-0102 Study, T. (2012). “Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks”.The Lancet 379 (9835): 2439–2448. doi:10.1016/S0140-6736(12)60917-9. PMID 22748591. edit
- Thaczuk, Derek and Carter, Michael. ICAAC: Best response to elvitegravir seen when used with T-20 and other active agents Aidsmap.com. 19 Sept. 2007.
The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
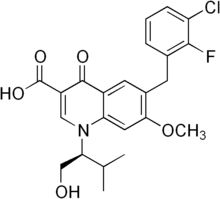
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:
WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid
THF
dichlorobis(triphenylphosphine)
palladium argon stream,
Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
-
Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium
Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.
chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
Elvitegravir dimer impurity, WO2011004389A2
Isolation of 1-[(2S)-1-({3-carboxy-6-(3-chloro-2-fluorobenzyl)-1 -[(2S)-I- hydroxy-3-methylbutan-2-yl]-4-oxo-1 , 4-dihydroquinolin-7-yl}oxy)-3- methylbutan-2-yl 6-(3-chloro-2-fluorobenzyl)-7-methoxy-4-oxo-1 , 4-dihydroquinoline-3-carboxylic acid (elvitegravir dimer impurity, 13)
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.
1H-NMR (DMSO-Cf6, 300 MHz, ppm) – δ 0.79 (m, d=6.3 Hz, 6H, 20 & 2O’)\ 1.18 & 1.20 (d, J=6.3 Hz & J=6.2 Hz, 6H, 21 & 21′)1, 2.42-2.49 (m, 2H, 19 & 19′), 3.81-3.89 (m, 3H, T & 17′Ha), 3.94-4.01 (m, 1 H, 17′Hb), 4.01 (s, 3H, 23), 4.11 (s, 2H, 7), 4.83-4.85 (m, 3H, 17 & 18′), 5.22 (t, J=4.7 Hz, 1H, OH), 5.41-5.44 (m, 1 H, 18), 6.73-6.78 (t, J=7.1 Hz, 1 H, 11)1‘ 2, 6.92-6.98 (t, J=8.0 Hz, 1H, 3′) 1‘2, 7.12-7.22 (m, 2H, 1 & 3), 7.34-7.39 (m, 1H, 2′),
7.45-7.48 (m, 1 H, 2), 7.49, 7.56 (s, 2H, 15 & 15′), 7.99, 8.02 (s, 2H, 9 & 9′), 8.89, 9.01 (s, 2H, 13 & 13′), 15.30, 15.33 (s, 2H, COOH’ & COOH”).
13C-NMR (DMSO-Cf6, 75 MHz, ppm)- δ 18.87, 19.03 (2OC, 20′C), 19.11 , 19.24 (21 C, 21 ‘C), 27.94 (7′C), 28.40 (7C), 28.91 , 30.08 (19C, 19′C), 56.80(23C), 60.11 (171C), 63.59 (18C), 66.52 (18′C), 68.53 (17C), 97.86, 98.97 (15, 15′), 107.43, 108.16 (12C, 12′C),
118.77, 119.38 (1OC, 10′C), 119.57 (d, J=17.6 Hz, 41C), 119.61 (d, J=17.9 Hz, 4C),
124.88 (d, J=4.3 Hz, 31C), 125.18 (d, J=4.2 Hz, 3C), 126.59, 126.96 (9C1 9′C), 127.14 (8′C), 127.62 (d, J=15.9 Hz, 61C), 127.73 (8C), 127.99 (d, J=15.2 Hz, 6C), 128.66 (2′C),
128.84 (11C), 128.84 (2C), 130.03 (d, J=3.4 Hz, 1C), 142.14, 142.44 (14C, 14′C), 144.37, 145.56 (13C, 131C), 155.24 (d, J=245.1 Hz, 5′C)1 155.61 (d, J=245.1 Hz, 5C),
160.17 (16′C), 162.04 (16C), 166.00, 166.14 (22C, 22′C), 176.17, 176.22 (11C, 111C).
DIP MS: m/z (%)- 863 [M+H]+, 885 [M+Na]+.
........................................
3.DOLUTEGRAVIR

Dolutegravir
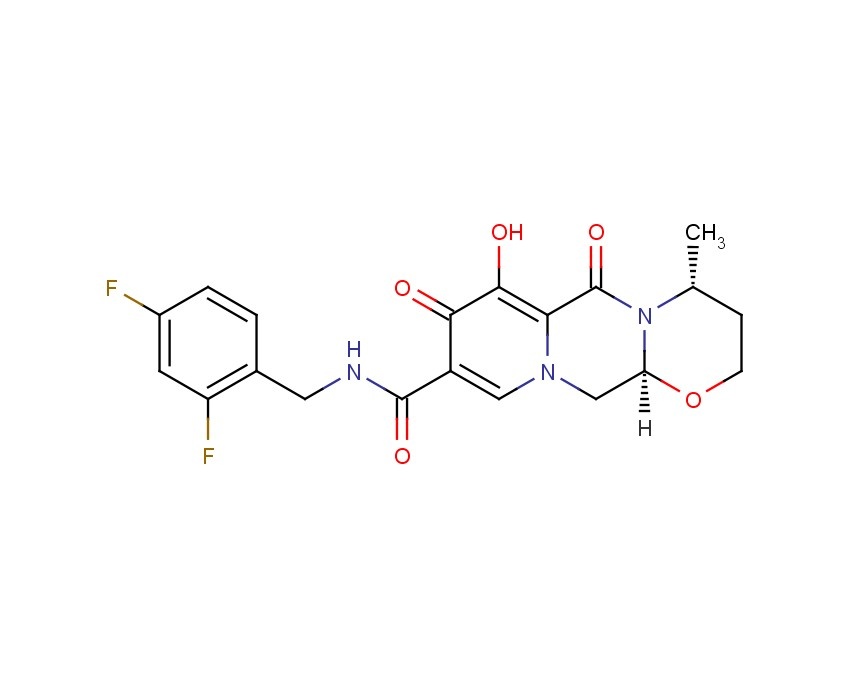
2H-Pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just "572" the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir's approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
..........................................
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
- Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

- This document describes the following reaction formula as a method of producing compound (I).


- Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
................................................
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
...............................................................
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)
...........................
SYNTHESIS
2H-Pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ...........dolutegravir
PATENT US8129385
Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt
a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt
The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES
IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
....................
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
- N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
- Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
- Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
- 62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
- Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
- Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
- Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N',N'-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
- 1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
- Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
- The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
- HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
- The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2

- Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
.................
ENTRY 68
...............................
WO 2010068262
..............................
WO 2010068253
.......................................
WO 2011119566
................................
Example 3
I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was - and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained - was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was - and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino - - butane - 1 - ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained - and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F - was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G - was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ', N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and '- tetramethyl-O-(yl 7 - aza - - benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 - was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained - was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was - and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino - - butane - 1 - ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained - and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F - was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G - was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ', N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and '- tetramethyl-O-(yl 7 - aza - - benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 - was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- "U.S. FDA approves GlaxoSmithKline's HIV drug Tivicay". Reuters. 12 August 2013. Retrieved 13 February 2013.
- "GSK wins priority status for new HIV drug in U.S". Reuters. 16 February 2013. Retrieved 18 February 2013.
- "ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV". Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline's HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- "Dolutegravir Prescribing Information". Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). "Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.". The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- Jump up^ http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). "Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection". New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. "SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA - See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf". Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). "Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.". The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds - [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
.....................
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html
4


GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
UNII-8GB79LOJ07; 8GB79LOJ07



BICTEGRAVIR
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
PATENTS
WO2014100323
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide

42

Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015177537&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196137&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
General Scheme I:

General Scheme II:



General Scheme II
General Scheme III:


General Scheme III
General Scheme IV:


G-1
General Scheme V:

II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:

[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:

A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:

E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA

C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:

K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
see..........http://newdrugapprovals.org/2015/12/28/gs-9883-bictegravir-an-hiv-1-integrase-inhibitor/
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
GS 9883, Bictegravir
GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
| MW | 449.37993 g/mol |
|---|
BICTEGRAVIR
- 16 Nov 2015 Phase-III clinical trials in HIV-1 infections (Combination therapy, Treatment-naive) in USA (PO) (Gilead Pipeline, November 2015)
- 01 Jul 2015 Gilead Sciences completes a phase I trial in HIV-1 infections in USA and New Zealand (NCT02400307)
- 01 Apr 2015 Phase-I clinical trials in HIV-1 infections (In volunteers) in New Zealand (PO) (NCT02400307)
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
SYNTHESIS COMING……..
PATENTS
WO2014100323
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
42
Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015177537&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196137&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide

-
1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesulfonic acid (0.195 mL, 3 mmol) and placed in a 75 deg C. bath. The reaction mixture was stirred for 7 h, cooled and stored at −10° C. for 3 days and reheated to 75° C. for an additional 2 h. This material was cooled and carried on crude to the next step.
-
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (1R,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85° C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HCl (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichlormethane:hexanes afforded the desired intermediate 42A: LCMS-ESI+ (m/z): [M+H]+ calculated for C15H17N2O6: 321.11; found: 321.3.
-
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)methaniminium hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LCMS-ESI+ (m/z): [M+H]+ calculated for C22H21F3N3O5: 464.14; found: 464.2.
-
To the crude reaction mixture of the previous step was added MgBr2 (0.258 g, 1.40 mmol). The reaction mixture was stirred at 50° C. for 10 minutes, acidified with 10% aqueous HCl, and extract twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN/H2O with 0.1% TFA modifier) to afford compound 42: 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.34 (t, J=5.7 Hz, 1H), 8.42 (s, 1H), 7.19 (t, J=8.7 Hz, 2H), 5.43 (dd, J=9.5, 4.1 Hz, 1H), 5.08 (s, 1H), 4.66 (dd, J=12.9, 4.0 Hz, 1H), 4.59 (s, 1H), 4.56-4.45 (m, 2H), 4.01 (dd, J=12.7, 9.7 Hz, 1H), 1.93 (s, 4H), 1.83 (d, J=12.0 Hz, 1H), 1.56 (dt, J=12.0, 3.4 Hz, 1H). LCMS-ESI+ (m/z): [M+H]+ calculated for C21H19F3N3O5: 450.13; found: 450.2.
PATENT
WO-2015195656General Scheme I:
General Scheme II:
General Scheme II
General Scheme III:
General Scheme III
General Scheme IV:
G-1
General Scheme V:
II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:
[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:
A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:
E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA
C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:
K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
| Patent | Submitted | Granted |
|---|---|---|
| POLYCYCLIC-CARBAMOYLPYRIDONE COMPOUNDS AND THEIR PHARMACEUTICAL USE [US2014221356] | 2013-12-19 | 2014-08-07 |
| US9216996 | Dec 19, 2013 | Dec 22, 2015 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
see..........http://newdrugapprovals.org/2015/12/28/gs-9883-bictegravir-an-hiv-1-integrase-inhibitor/
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
Share this:
.........................
5
This comment has been removed by the author.
ReplyDeleteHello everyone in this forum i am LEOKADIA STELLA from USA. i was waiting for this days to come so that i will share a testimony of how i was cure of my hiv/aids by DR ODUWA the great healer. i will never forget this two days in my life the day i tested hiv positive and the day i tested negative. the day i tested positive was the saddest day for me but the day i tested negative was the happiest day for me, but who was the source of my happiness is God and DR ODUWA the great healer, that is the reason i am taking all my time to share this testimony so that people who might also need help of curing their sickness to contact him through his email: dr.oduwaspellhome@gmail.com as i earlier did when i was searching for help. This man brought countless joy into my life and my family as well. Well i have really believe that God sent you to heal his people from this deadly disease. i will forever give thanks to you for healing me from hiv. Please kindly contact this great healer if you need his help because he can save your soul he just saved mine. Contact email address: dr.oduwaspellhome@gmail.com
DeleteThe article is very informative.B2B Contact List is one of the leading database vendors b2bcontactliststhat provide an accurate email list of various categories
ReplyDeletelike IT, Healthcare, Industries globally.We provide you a list of decision-makers at your fingertips where you can
shorten your sales cycle and increase your revenue. Chemical Drug Dependency Counselors Mailing Lists
The article is very informative.B2B Contact List is one of the leading database vendors b2bcontactliststhat provide an accurate email list of various categories
ReplyDeletelike IT, Healthcare, Industries globally.We provide you a list of decision-makers at your fingertips where you can
shorten your sales cycle and increase your revenue. Chemical Drug Dependency Counselors Email Lists
Hello everyone in this forum i am LEOKADIA STELLA from USA. i was waiting for this days to come so that i will share a testimony of how i was cure of my hiv/aids by DR ODUWA the great healer. i will never forget this two days in my life the day i tested hiv positive and the day i tested negative. the day i tested positive was the saddest day for me but the day i tested negative was the happiest day for me, but who was the source of my happiness is God and DR ODUWA the great healer, that is the reason i am taking all my time to share this testimony so that people who might also need help of curing their sickness to contact him through his email: dr.oduwaspellhome@gmail.com as i earlier did when i was searching for help. This man brought countless joy into my life and my family as well. Well i have really believe that God sent you to heal his people from this deadly disease. i will forever give thanks to you for healing me from hiv. Please kindly contact this great healer if you need his help because he can save your soul he just saved mine. Contact email address: dr.oduwaspellhome@gmail.com
ReplyDeleteNowadays, pills can cure a lot of diseases! There are pills that can help fight HIV diseases. Tablets quality and work like a clock!
ReplyDeleteFind out more here https://prepgeneric.com/catalog/isentress/
This comment has been removed by the author.
ReplyDelete