1 RAMOSETRON
2 DOLASETRON
3 Ondansetron
4 GRANISETRON
5 ALOSETRON
6 TROPISETRON
7 GALDANSETRON
8 AZASETRON
ondansetron as the hydrochloride dihydrate, granisetron as the hydrochloride, dolasetron as the mesylate, tropisetron as the monohydrochloride, ramosetron, fabesetron, alosetron
and cilansetron as the hydrochlorides,
palonosetron as the monohydrochloride,
azasetron as the hydrochloride,
and zatosetron as the maleate
2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-1 H-ιmιdazol-4-yl)methyl]-1 H-pyr ιdo[4,3- b]ιndol-1 -one (see also alosetron, EP 0 306 323
(+)-10-methyl-7-(5-methyl-1 H-ιmιdazol-4-ylmethyl)-6,7,8,9-tetrahydropyπdo[1 ,2- a]ιndol-6-one (see also fabesetron, EP 0 361 317);
Ondansetron: 1,2,3 ,9-Tetrahydro-9-methyl-3-[(2-methyl1-H-imidazole-1-yl)methyl]-4H-carbazol-4-one

Granisetron: Endo-1-methyl-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)-1H-indazole-3-carboxamide

Tropisetron: Endo-1H-indole-3-carbocylic acid8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester

Dolasetron: 1H-Indole-3 -carboxylic acid (2a, 6a, 8a, 9up)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl Ester

Azasetron: (±)-N-Azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide

Alosetron: 2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl- 1H-imidazol-4-yl)methyl]-1H-pyrido[4,3-b]indol-1-one

Ramosetron


1 RAMOSETRON

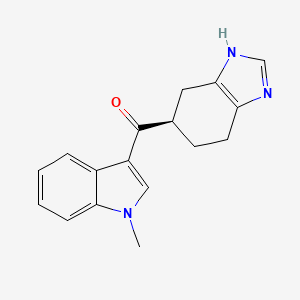
Ramosetron (INN),(1-methylindol-3-yl)-[(5R)-4,5,6,7-tetrahydro-3H-benzimidazol-5-yl]methanone, 132036-88-5 cas no
| C17H17N3O | |
| 279.33 g/mol |
(1-methyl-1H-indol-3-yl)[(5R)-4,5,6,7-tetrahydro-1H-benzimidazol-5-yl]methanone
YM060
- Nasea
- Nor-YM 060
- Ramosetron
- UNII-7ZRO0SC54Y
…………………………………………………………………………………..
HYDROCHLORIDE SALT
hyrochloride salt, cas no 132907-72-3
C17-H17-N3-O.Cl-H
315.8022
Yamanouchi (Originator)
GASTROINTESTINAL DRUGS, Irritable Bowel Syndrome, Agents for, Nausea and Vomiting, Treatment of, NEUROLOGIC DRUGS, 5-HT3 Antagonists
Launched-1996 JAPAN
(−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole monohydrochloride (yield 78.8%, 99.5% e.e.). FAB-MS (m/z): 280 [M+H+]
1H NMR (DMSO-d6, 30° C.): δ ppm (TMS internal standard): 1.82-1.95 (1H, m), 2.12-2.22 (1H, m), 2.66-2.94 (4H, m), 3.63-3.72 (1H, m), 3.88 (3H, s), 7.24 (1H, t, J=8.0 Hz), 7.30 (1H, t, J=8.0 Hz), 7.56 (1H, d, J=8.0 Hz), 8.22 (1H, d, J=8.0 Hz), 8.53 (1H, s), 8.90 (1H, s), 14.42 (1H, br)
…………………………………………………………………………………….
Ramosetron (INN) is a serotonin 5-HT3 receptor antagonist for the treatment of nausea and vomiting.[1] Ramosetron is also indicated for a treatment of “diarrhea-predominant irritable bowel syndrome in males”.[2] In India it is marketed under the brand name of“IBset”.
It is only licensed for use in Japan and selected Southeast Asian countries. In Japan it is sold under the tradename Iribo (イリボー). [3] Elsewhere it is commonly sold under the tradename Nasea and in India as Nozia (300 mcg/ml Inj. & 100 mcg Tab.) [4]
It is only licensed for use in Japan and selected Southeast Asian countries. In Japan it is sold under the tradename Iribo (イリボー). [3] Elsewhere it is commonly sold under the tradename Nasea and in India as Nozia (300 mcg/ml Inj. & 100 mcg Tab.) [4]
- Fujii Y, Saitoh Y, Tanaka H, Toyooka H (February 2000). “Ramosetron for preventing postoperative nausea and vomiting in women undergoing gynecological surgery”.Anesth. Analg. 90 (2): 472–5. doi:10.1097/00000539-200002000-00043.PMID 10648342.
- http://www.astellas.com/en/corporate/news/detail/astellas-launches-irribow-for.html
- Summary in Japanese. Retrieved on September 4, 2012.
- Abridged prescribing information – Nasea (MIMS Philippines). Retrieved on June 13, 2008.
- Synthesis and 5-HT3 antagonistic activities of 4,5,6, 7-tetrahydrobenzimidazole derivatives
200th ACS Natl Meet (August 26-31, Washington DC) 1990, Abst MEDI 39
| US7652052 | 1-27-2010 | Process for producing ramosetron or its salt |
| US5576014 | 11-20-1996 | Intrabuccally dissolving compressed moldings and production process thereof |
| US5496942 | 3-6-1996 | 5-substituted tetrahydrobenzimidazole compounds |
| US5466464 | 11-15-1995 | Intrabuccally disintegrating preparation and production thereof |
| US5344927 | 9-7-1994 | Tetrahydrobenzimidazole derivatives and pharmaceutical compositions containing same |
| WO9413284 | 6-24-1994 | NEW USE OF 5-HT3 RECEPTOR ANTAGONISTS |
AU 9048890; EP 0381422; JP 1991223278; US 5344927
| CN1696128A | Nov 2, 2004 | Nov 16, 2005 | 天津康鸿医药科技发展有限公司 | New method for synthesizing Ramosetron Hydrochloride |
| CN1765896A | Oct 28, 2004 | May 3, 2006 | 北京博尔达生物技术开发有限公司 | Novel preparation method of ramosetron hydrochloride |
| US5496942 * | 14 Feb 1994 | 5 Mar 1996 | Yamanouchi Pharmaceutical Co., Ltd. | 5-substituted tetrahydrobenzimidazole compounds |
| US5677326 * | 30 Sep 1994 | 14 Oct 1997 | Tokyo Tanabe Company Limited | Indoline compound and 5-HT.sub.3 receptor antagonist containing the same as active ingredient |
| US7358270 | 28 Jan 2005 | 15 Apr 2008 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7683090 | 18 Oct 2006 | 23 Mar 2010 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7794748 | 27 Aug 2004 | 14 Sep 2010 | Yamanouchi Pharmaceutical Co., Ltd. | Stable oral solid drug composition |
WO 2010024306
WO 2013005760
WO 2013100701
WO 2011001954
The chemical name of ramosetron is (−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole, and it has the structure represented by the formula (II).

It is known that ramosetron or a salt thereof has a potent 5-HT3 receptor antagonism (Patent Reference 1, Non-patent references 1 and 2), and it is on the market as a preventive or therapeutic agent for digestive symptoms (nausea, emesis) caused by administration of an anti-malignant tumor agent (cisplatin or the like). In addition, a possibility has been reported that ramosetron or a salt thereof may be useful as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome (Patent Reference 1), and its clinical trials are now in progress as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome.
As a process for producing ramosetron or a salt thereof, the following production methods are known.
Patent Reference 1 describes a production method shown by the following Production method A, namely a method for producing a tetrahydrobenzimidazole derivative (V) by allowing a heterocyclic compound (III) to react with a carboxylic acid represented by a formula (IV) or its reactive derivative.
(Production Method A)

(In the formula, X2 is a single bond and binds to a carbon atom on the heterocyclic ring represented by Het.)
As an illustrative production method of ramosetron, Patent Reference 1 describes a production method (Production method A-1) in which racemic ramosetron are obtained by using 1-methyl-1H-indole as the compound (III), and N,N-diethyl-4,5,6,7-tetrahydrobenzimidazole-5-carboxamide or N-[(4,5,6,7-tetrahydrobenzimidazol-5-yl)carbonyl]pyrrolidine, which are acid amides, as the reactive derivative of compound (IV), and allowing them to undergo treatment with phosphorus oxychloride (Vilsmeyer reaction), and then their optical resolution is carried out by fractional crystallization using (+)-dibenzoyltartaric acid.
In addition, the Patent Reference 1 exemplifies an acid halide as one of the reactive derivatives of the compound (IV), and also describes another production method of the compound (V) (Production method A-2) in which the heterocyclic compound (III) is condensed with an acid halide of the compound (IV) by the Friedel-Crafts acylation reaction using a Lewis acid as the catalyst. However, illustrative production example of ramosetron by the Friedel-Crafts acylation reaction is not described therein.
Also, a method similar to the Production example A-1 is described in Non-patent References 1 and 2 as a production method of ramosetron.
In addition, Non-patent Reference 3 describes a method for producing ramosetron labeled with 11C, represented by a Production method B. However, it discloses only the methylation step, and does not disclose a production method of nor-YM060 as the starting material.
(Production Method B)

(In the formula, nor-YM060 means (R)-5-[(1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole which was provided by the present applicant, DMF means dimethylformamide.)
- Non-patent Reference 1: Chemical & Pharmaceutical Bulletin, 1996, vol. 44, no. 9, p. 1707-1716
- Non-patent Reference 2: Drugs of the Future, 1992, vol. 17, no. 1, p. 28-29
- Non-patent Reference 3: Applied Radiation and Isotopes, 1995, vol. 46, no. 9, p. 907-910
- Patent Reference 1: JP-B-6-25153

LIU Qing-wen, XU Hao, TIAN Hua, ZHENG Liang-yu, ZHANG Suo-qin
Chemoenzymatic Synthesis of Ramosetron Hydrochloride
Chemoenzymatic Synthesis of Ramosetron Hydrochloride
2012 Vol. 28 (1): 70-72 [Abstract] ( 1143 ) [HTML 1KB] [PDF 206KB] ( 1052 )
doi:http://www.cjcu.jlu.edu.cn/hxyj/EN/abstract/abstract13356.shtml
doi:http://www.cjcu.jlu.edu.cn/hxyj/EN/abstract/abstract13356.shtml
…………………………………………………………………………..

The Vilsmeier-type reaction of 1-methylindole (I) with 5 – (1-pyrrolidinocarbonyl) -4,5,6,7-1 H-tetrahydrobenzimidazole hydrochloride (II) and phosphorous oxychloride in 1,2-dichloroethane gives (-5? -. [(1-methyl-3-indolyl) carbonyl] -4,5,6,7-tetrahydro-1H-benzimidazol e (III) Optical resolution of (III) with (+)-dibenzoyltartaric acid (DIBTA) in DMF -H2O, followed by exchange of the salt affords YM060.
………………………………………………….
2 DOLASETRON

DOLASETRON
(3R)-10-oxo-8-azatricyclo[5.3.1.03,8]undec-5-yl 1H-indole-3-carboxylate, 115956-12-2 CAS

Dolasetron (trade name Anzemet) is a serotonin 5-HT3 receptor antagonist used to treat nausea and vomiting following chemotherapy. Its main effect is to reduce the activity of the vagus nerve, which is a nerve that activates the vomiting center in the medulla oblongata. It does not have muchantiemetic effect when symptoms are due to motion sickness. This drug does not have any effect on dopamine receptors or muscarinic receptors.
Dolasetron breaks down slowly, staying in the body for a long time. One dose usually lasts 4 to 9 hours and is usually administered once or twice daily. This drug is removed from the body by the liver and kidneys.
- Chemotherapy-induced nausea and vomiting
- 5-HT3 receptor antagonists are the primary drugs used to treat and prevent chemotherapy-induced nausea and vomiting. Many times they are given intravenously about 30 minutes before beginning therapy.
- Post-operative and post-radiation nausea and vomiting
- Is a possible therapy for nausea and vomiting due to acute or chronic medical illness or acute gastroenteritis
- Unlike most other 5HT3 antagonists, data is lacking for use of dolasetron with aprepitant in chemotherapy-induced nausea and vomiting (CINV).
- It is also sometimes used as an antiemetic (anti-vomiting medication) in veterinary medicine for dogs and cats.
Dolasetron is a well-tolerated drug with few side effects. Headache, dizziness, and constipation are the most commonly reported side effects associated with its use. There is a potential for prolonging of the QT interval to occur as well. There have been no significant drug interactions reported with this drug's use. It is broken down by the liver's cytochrome P450 system and it has little effect on the metabolism of other drugs broken down by this system. Intravenous dolasetron is contraindicated in Chemotherapy-induced nausea and vomiting (CINV). Doxorubicin and cyclophosphamide are as emetogenic as cisplatin, and preventive drugs should always be considered. The 5HT3 agonists are the mainstays of prevention and are frequently used in combination with other drugs such as corticosteroids and the NK1 receptor antagonist aprepitant. However, the FDA recently issued a drug communication stating that the injection form of dolasetron, a 5HT3 agonist, should no longer be used in adult or pediatric patients with CINV. Dolasetron injection can increase the risk of developing torsade de pointes, a potentially fatal abnormal heart rhythm. Patients with underlying heart conditions or existing heart rate or rhythm problems are at increased risk. Although the oral form of this agent can still be used, careful monitoring and correction of potassium and magnesium levels should be initiated prior to and during treatment. In addition, in older patients and in patients with heart failure, a slow heart rate, underlying cardiac disease, and those with renal impairment, monitoring with electrocardiography is indicated when this drug is used. Congenital long-QT syndrome and drugs that prolong the PR or QRS interval are contraindications to dolasetron therapy. Dolasetron injection may still be used for the prevention and treatment of postoperative nausea and vomiting. As per Food and Drug Administration.
- http://www.fda.gov/Drugs/DrugSafety/ucm237081.htm
- Katzung, Bertram G. Basic and Clinical Pharmacology, 9th ed. (2004). ISBN 0-07-141092-9
.............

ANZEMET (dolasetron mesylate) is an antinauseant and antiemetic agent. Chemically, dolasetron mesylate is (2α,6α,8α,9aβ)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl-lH- indole-3-carboxylate monomethanesulfonate, monohydrate. It is a highly specific and selective serotonin subtype 3 (5-HT3) receptor antagonist both in vitro and in vivo. Dolasetron mesylate has the following structural formula:
|
The empirical formula is C19H20N2O3 • CH3SO3H • H2O, with a molecular weight of 438.50.
Approximately 74% of dolasetron mesylate monohydrate is dolasetron base.
Dolasetron mesylate monohydrate is a white to off-white powder that is freely soluble in water and propylene glycol, slightly soluble in ethanol, and slightly soluble in normal saline.
ANZEMET Injection (dolasetron mesylate injection) is a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each milliliter of ANZEMET Injection (dolasetron mesylate injection) contains 20 mg of dolasetron mesylate and 38.2 mg mannitol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.8.
ANZEMET Injection (dolasetron mesylate injection) multidose vials contain a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each ANZEMET multidose vial contains 25 mL (500 mg) dolasetron mesylate. Each milliliter contains 20 mg dolasetron mesylate, 29 mg mannitol, USP, and 5 mg phenol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.7.
Synthesis of Dolasetron base is not very widely reported in literature. However, EP0266730/U.S. Pat. No. 4,906,755 describes process for the preparation endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate, by the condensation of diethyl malonate with cis-1,4-dichloro-2-butene (2) in presence of lithium hydride in dimethylformamide to give diethyl-3-cyclopentene-1,1-dicarboxylate (3), which on hydrolysis and decarboxylation gave 3-cyclopentene-1-carboxylic acid (4). The compound (4) was further treated with thionyl chloride and pyridine in ethanol to obtain ethyl 3-cyclopentene-1-carboxylate (5). Compound (5) was oxidized to 4-ethoxycarbonyl-1,2-cyclopentanediol (6) by using N-methylmorpholine N-oxide in the presence of osmium tetroxide catalyst. The diol (6) was cleaved to the β-ethoxycarbonylglutaraldehyde (7) using sodium periodate and used directly in the next reaction. Robinson-Schopf cyclisation of the compound (7) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride resulted in the pseudopelletierine derivative i.e. 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-one (8). The ketone group of compound (8) was reduced with sodiumborohydride in ethanol to give 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (9). The reduced alcohol (9) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (10). Dieckmann cyclisation of the compound (10) using strong base (potassium t-butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the desired alcohol. The resulting alcohols can exist in two conformations—axial and equatorial. The main product obtained by above procedure was the axial alcohol or endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) and it can be separated from the equatorial isomer by crystallization of the camphorsulfonate or tetrafluoroborate salt. The tetrafluoroborate salt of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) was further reacted with 3-indolecarboxylic acid chloride in presence of silver tetrafluoroborate in anhydrous nitroethane at −78° C. to endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base, which was further converted into Dolasetron mesylate monohydrate (Scheme I) with a yield of 66%. No further purification is described.
The above process uses column chromatography for purification of compounds (9) and (10), which is expensive, time consuming and impractical on an industrial scale. The above patent does not disclose the yield and purity of Dolasetron mesylate obtained and so also for the intermediates. In addition, Osmium tetroxide used for preparation of compound (6) is toxic, has a corrosive action on eyes and hence difficult to use at industrial scale. Also this process uses high volume of water during preparation of the compound (8); preparation of compound (II) from compound (10) is tedious, because the workup involves several extractions with ethyl acetate and preparation of compound (I) in presence of silver tetrafluoroborate involves the use of expensive silver compound.

Another method described in EP0339669 provides a process for the preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of dimethyl malonate with cis-1,4-dichloro-2-butene (2) in presence of lithium hydride in dimethyl formamide to give dimethyl-3-cyclopentene-1,1-dicarboxylate (12), which was decarbomethylated to obtain methyl-3-cyclopentene-1-carboxylate (13). This compound (13) was treated with m-chloroperbenzoic acid in dichloromethane to obtain 1-methoxycarbonyl-3-cyclopenteneoxide (14). The compound (13) on ozonolysis gave β-methoxycarbonylglutaraldehyde (15) or the epoxide (14) was reacted with periodic acid to obtain the β-methoxycarbonylglutaraldehyde (15), which was used directly in the next reaction. Robinson-Schopf cyclisation of the compound (15) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride gave the pseudopelletierine derivative i.e. 7-methoxycarbonyl-9-(methoxycarbonylethyl)-9-azabicyclo[3.3.1]nonan-3-one (16). The ketone group of compound (16) was reduced with sodiumborohydride in methanol to give 7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (17). The reduced alcohol (17) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (18a) or treated with methylal to protect the hydroxyl group to obtain 3-methoxymethoxy-7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo[3.3.1]nonan-3-ol (18b).
Dieckmann cyclisation of the compound (18) using strong base (potassium t-butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11). The alcohol (11) was further reacted with 3-indolecarboxylic acid in presence of trifluoroacetic anhydride in dichloromethane to endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base (A), which was then converted into Dolasetron mesylate (1) (not shown in Scheme II) by treating with methanesulphonicacid in acetone (Scheme II).

Disadvantages of this process are:
- (i) use of high volume of water for preparation of compound (16) and
- (ii) preparation of compound (11) from compound (18) which is tedious because at the time of workup, ethyl acetate extractions take up longer period (20 h).
The process is not only time consuming but also expensive on an industrial scale. The patent does not disclose purity of Dolasetron base obtained nor for any of the intermediates.
The process as described in EP 0266730 involves treatment of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one (Dolasetron base) with a solution of methane sulfonic acid in ethanol to provide Dolasetron mesylate monohydrate. EP 0339669 describes crystallization of crude Dolasetron mesylate by dissolution in aqueous isopropanol and regeneration by adding ether. The polymorphic form obtained by the processes described in U.S. Pat. No. 4,906,755/EP 0266730 and EP 0339669 is designated herein as Dolasetron mesylate Form I.
The ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice is called polymorphism. Many organic compounds including active pharmaceutical ingredients (API's) exhibit polymorphism.
Drug substance existing in various polymorphic forms differs from each other in terms of stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
Rate of dissolution of an API's in patient's stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administrated API can reach the patient bloodstream. Flowability affects the ease with which the material is handled while processing a pharmaceutical product.
Investigation of crystal polymorphism is an essential step in pharmaceutical research due to the influence of solid-state properties on dosage form.
As the polymorphs are known to possess different spectroscopic properties, technique such as X-Ray powder diffraction (XRPD), Fourier transformer Infrared (FT-IR) spectroscopy, Solid State 13C-NMR, and thermal method of analysis are keys to identify and characterize the new polymorphs or existing polymorphs.
The discovery of new polymorphs with same or better pharmaceutical equivalence and bioequivalence as that of the known polymorphs provides an opportunity to improve the performance characteristic of the pharmaceutical product.
The prior art describes isolation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base as an oil. It is desirable to have the product in the solid form than oil, as solid is easy to handle and easy to purify.
Dolasetron base is isolated as a solid in EP 0339669. However, there is no evidence of polymorphism.
WO2006056081 discloses purification of Dolasetron base using strong acid especially methanesulphonic acid in presence of acid halide.
3 ONDANSETRON
Ondansetron

 ondansetron
ondansetron
Ondansetron hydrochloride dihydrate, cas 99614-01-4, GG-032, SN-307, GR-C505/75,
C18-H19-N3-O.Cl-H.2-H2-O
365.8586
GlaxoSmithKline (Originator), Sankyo (Licensee), Vita (Licensee)
Nausea and Vomiting, Treatment of, NEUROLOGIC DRUGS, 5-HT3 Antagonists
Launched-1990
Ondansetron hydrochloride dihydrate is a serotonin-3 (5-HT3) receptor antagonist. J Org Chem1980, 45, (15): 2938 Heterocycles1997, 45, (10): 2041 EP 0595111 WO 0172716, Ondansetron (INN) (/ɒnˈdænsɛtrɒn/; developed and first marketed by GlaxoSmithKline as Zofran) is a serotonin 5-HT3 receptor antagonist used mainly as an antiemetic (to treat nausea and vomiting), often following chemotherapy. It affects both peripheral and central nerves. Ondansetron reduces the activity of the vagus nerve, which deactivates the vomiting center in the medulla oblongata, and also blocks serotonin receptors in thechemoreceptor trigger zone. It has little effect on vomiting caused by motion sickness, and does not have any effect on dopamine receptors ormuscarinic receptors. Although an effective anti-emetic agent, the high cost of brand-name ondansetron initially limited its use to controlling postoperative nausea and vomiting (PONV) and chemotherapy-induced nausea and vomiting (CINV). The active ingredient in ZOFRAN Tablets and ZOFRAN Oral Solution is ondansetron hydrochloride (HC1) as the dihydrate, the racemic form of ondansetron and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (±) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-l-yl)methyl]-4H-carbazol-4-one, monohydrochloride, dihydrate. It has the following structural formula:
 |
The empirical formula is C18H19N3O•HCl•2H2O, representing a molecular weight of 365.9. Ondansetron HC1 dihydrate is a white to off-white powder that is soluble in water and normal saline. The active ingredient in ZOFRAN ODT Orally Disintegrating Tablets is ondansetron base, the racemic form of ondansetron, and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (+) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-l-yl)methyl]-4H-carbazol-4-one. It has the following structural formula:
 |
The empirical formula is C18H19N3O representing a molecular weight of 293.4. The 5-HT3 receptor antagonists are the primary drugs used to treat and prevent chemotherapy-induced nausea and vomiting (CINV). A common use case is to give them intravenously about 30 minutes before commencement of a chemotherapy treatment.
Ondansetron is used off-label to treat morning sickness and hyperemesis gravidarum of pregnancy. A cohort study of over 600,000 pregnancies in Denmark found that ondansetron administration during pregnancy is not associated with a significantly increased risk of spontaneous abortion,stillbirth, major birth defect, preterm birth, low birth weight, or small for gestational age. However, in practice, ondansetron is typically used after trials of other drugs have failed.
Ondansetron is one of several anti-emetic agents used during the vomiting phase of cyclic vomiting syndrome. Trials in emergency department (ED) settings support the use of ondansetron to reduce vomiting associated with gastroenteritis and dehydration.A retrospective review found that it was used commonly for this purpose, being administered in over 58% of cases. Its use reduced hospital admissions, but was also associated with higher rates of return visits to the ED. Furthermore, patients who had initially received ondansetron were more likely to be admitted on the return visit than patients who had not received the drug. However, this effect may simply be due to the agent being used more frequently in patients who present with more severe illness. Its use was not found to mask serious diagnoses.
.JPG)
A vial of ondansetron for intravenous injection
Ondansetron was developed around 1984 by scientists working at Glaxo’s laboratories in London. It is in both the imidazole and carbazole families of heterocyclic compounds. After several attempts the company successfully filed for U.S. patent protection for the drug in 1986 and was granted in June 1988 while a use patent was granted in June 1988. A divisional use patent was granted on November 26, 1996. Ondansetron was granted FDA approval as Zofran in January 1991. Glaxo did pediatric research on Zofran’s uses, and gained a patent extension as a result, extending U.S. exclusivity until December 24, 2006. The FDA subsequently approved the first generic versions in December 2006, with marketing approval granted to Teva Pharmaceuticals USA and SICOR Pharmaceuticals.
Ondansetron is marketed by GlaxoSmithKline (GSK) under the trade name Zofran. Other manufacturers include Pfizer Injectables (Ondanzetron), Opsonin Pharma Bangladesh (Anset), Strativa Pharmaceuticals (Zuplenz), Indswift Ltd. (Ondisolv), Cipla Ltd. (Emeset), Gedeon Richter Ltd. (Emetron), Korea United Pharmaceuticals (Emodan), Zentiva a.s. (Ondemet), Strides Arcolab (Setronax), Emistat (Unimed and Unihealth Bangladesh Ltd.)Glenmark Generics Ltd. (India) (Ondansetron) and Novell Pharmaceutical Laboratories (Ondavell). On May 29, 2006, Baxter Healthcare received tentative approval to market its own label of Ondansetron Injection, USP, 8 mg/50 mL and 32 mg/50 mL iso-osmotic sodium chloride solution, beginning upon expiration of GSK’s patent later that year.
In 1997, ondansetron was the subject of a meta-analysis case-study published in the British Medical Journal. Researchers examined 84 trials, with 11,980 patients receiving ondansetron, published between 1991 and September 1996. Intravenous ondansetron 4 mg versus placebo was investigated in 16 reports and three further reports which had been duplicated six times. The number needed to treat (NNT) to prevent vomiting within 24 hours was 9.5, with 95% confidence interval 6.9 to 15, in the 16 non-duplicated reports. In the three duplicated reports, the NNT was significantly lower at 3.9 (3.3 to 4.8) with P<0.00001. When all 25 reports were combined the apparent number needed to treat improved to 4.9 (4.4 to 5.6). Inclusion of duplicate reports led to a 23% overestimation of ondansetron’s antiemetic efficacy. In addition, the authors found that the covert duplication of reports on ondansetron was not easy to detect, because of lack of cross-referencing between papers, and that reports containing duplicate findings were cited in eight reviews of the drug. Their analysis was a subject of an editorial in the Journal of the American Medical Association in 1999.………………………
patents
AU 8538097; BE 0901576; CH 664152; ES 8609309; ES 8708224; ES 8801247; FR 2561244; GB 2153821; JP 1985214784
……………..
(±) l,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-lh-imidazol-l-yl)methyl]-4h- carbazol-4-one having the molecular structure

is a selective 5-HT3 receptor antagonist. It is known by the generic nameondansetron. Ondansetron reduces nausea in patients undergoing chemotherapy. Grunberg, S.M.; Hesketh, P.J. “Control of Chemotherapy-Induced emesis” N. Engl. J. Med. 1993, 329, 1790-96. Ondansetron is indicated for prevention of nausea and vomiting associated with some cancer chemotherapy, radiotherapy and postoperative nausea and/or vomiting.
Several chemical processes are known from the literature for the synthesis ofondansetron. GB-Pat. 2 153 821 and 2 192 885 describe syntheses starting from carbazolone derivative, and EP-Pat. 595 111 as well as a Hungarian patent application ( P 00-01287 ) give detailed information about some different chemical procedures.
Ondansetron is currently available as an anti-emetic agent, particularly in cancer chemotherapy, and in some other uses such as anti-depressive, anti- migraine and anti-psychotic. It is commonly used in the alleviation of cognitive disorders as in Alzheimer disease, in treatment of rhinitis, psychiatric disorders and for increased vigilance and for control of dependence on narcotics.
U.S. Patent No. 4,695,578, assigned to the Glaxo Group Limited, describes a process of preparing ondansetron and uses thereof. However, ondansetronprepared according to said process contains impurities and by-products such as l,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one.
The hydrochloride salt of ondansetron is generally safe for oral administration to a patient without causing irritation or other adverse effect. The hydrochloride salt is marketed in tablet form and in oral solution form under the brand name Zofran®. The tablet’s active ingredient is a dihydrate of ondansetronhydrochloride containing two molecules of bound water in ondansetronhydrochloride’ s crystal lattice. The present invention relates to the solid state physical properties of ondansetron hydrochloride. These properties can be influenced by controlling the conditions under which the hydrochloride salt is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
These important physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic form of a substance. Llacer and coworkers have postulated that different spectroscopic characteristics of samples of ondansetron free base prepared differently could be attributable to two different configurations about the methylene bridge between the 1, 2, 3, 9-tetrahydrocarbazol-4-one ring and the imidazole ring. Llacer, J.M.; Gallardo, V.; Parera, A. Ruiz, M.A. InternJ.Pharm., 177, 1999, 221-229.
(±)1,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-1h-imidazol-1-yl)methyl]-4h-carbazol-4-one having the molecular structure

is a selective 5-HT3 receptor antagonist. It is known by the generic nameondansetron. Ondansetron reduces nausea in patients undergoing chemotherapy. Grunberg, S. M.; Hesketh, P. J. “Control of Chemotherapy-Induced emesis” N. Engl. J. Med. 1993, 329, 1790-96. Ondansetron is indicated for prevention of nausea and vomiting associated with some cancer chemotherapy, radiotherapy and postoperative nausea and/or vomiting.
The hydrochloride salt of ondansetron is generally safe for oral administration to a patient without causing irritation or other adverse effect. The hydrochloride salt is marketed in tablet form and in oral solution form under the brand name Zofran®. The tablet’s active ingredient is a dihydrate of ondansetronhydrochloride containing two molecules of bound water in ondansetronhydrochloride’s crystal lattice.
The present invention relates to the solid state physical properties ofondansetron hydrochloride. These properties can be influenced by controlling the conditions under which the hydrochloride salt is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally-administered active ingredient can reach the patient’s bloodstream. The rate of dissolution is also a consideration in formulating syrups, elixirs and other liquid medicaments. The solid state form of a compound may also affect its behavior on compaction and its storage stability.
These important physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic form of a substance. Llacer and coworkers have postulated that different spectroscopic characteristics of samples ofondansetron free base prepared differently could be attributable to two different configurations about the methylene bridge between the 1,2,3,9-tetrahydrocarbazol-4-one ring and the imidazole ring. Llacer, J. M.; Gallardo, V.; Parera, A. Ruiz, M. A. Intern.J.Pharm., 177, 1999, 221-229.
A crystalline polymorphic form of a compound may exhibit different thermal behavior from amorphous material or another polymorphic form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and can be used to distinguish some polymorphic forms from others. A particular polymorphic form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state 13C NMR spectrometry and infrared spectrometry. There is a wide variety of techniques that have the potential of producing different crystalline forms of a compound. Examples include crystallization, crystal digestion, sublimation and thermal treatment.
U.S. Pat. No. 4,695,578, Example 1a, discloses a preparation ofondansetron by alkylation of 2-methylimidazole with 2,3,4,9 tetrahydro-N,N,N,9-tetramethyl-4-oxo-1H-carbazole-3-methanaminium iodide. In this example,ondansetron was isolated as its hydrochloride salt by suspending the reaction product in a mixture of absolute ethanol and ethanolic HCl, warming the suspension, filtering to remove impurities and precipitating the hydrochloride salt with dry ether.
In Example 10 of the ’578 patent, ondansetron free base was converted into a hydrochloride salt dihydrate by dissolving the free base in a mixture of isopropanol and water and treating it with concentrated hydrochloric acid. After filtration at elevated temperature, ondansetron was driven out of solution by adding additional isopropanol and cooling. The dihydrate was obtained as a white crystalline solid by recrystallizing it from a 6:10 mixture of water and isopropanol. Ondansetron hydrochloride dihydrate obtained by following Example 10 of the ’578 patent is denominated Form A in this disclosure. Powdered samples of Form A produce a powder X-ray diffraction pattern essentially the same as the pattern shown in FIG. 1.
U.S. Pat. No. 5,344,658 describes ondansetron having a particular particle size distribution and the use of such ondansetron in a pharmaceutical composition. The particle size of ondansetron hydrochloride dihydrate obtained by crystallization from a solvent is reduced by desolvating them, e.g. by heating, and then exposing the desolvated crystals to a humid atmosphere. A collection of crystals obtained by this particle size reduction process is said to consist exclusively of crystals of less than 250 micron size and to contain 80% or more crystals of less than 63 microns. Crytals size was determined by air jet seive analysis.
According to the ’658 patent, ondansetron hydrochloride dehydrate having the same particle size distribution as the rehydrated ondansetron hydrochloride also is provided as part of that invention. Since only one process for dehydratingondansetron hydrochloride is described in the ’658 patent, a dehydrate is evidently the intermediate compound that is rehydrated in the particle size reduction process.
U.S. Pat. Nos. 4,695,578 and 5,344,658 are incorporated herein by reference.
U.S. Pat. No. 4,695,578 (’578 patent) discloses a process for preparingondansetron hydrochloride dihydrate having a large particle size (e.g., less than about 60% of the particles are smaller than 250 μm). The ’578 patent process involves the step of cooling a solution of ondansetron hydrochloride, isopropanol, and water, optionally followed by an additional step of recrystallizing from a mixture of water and isopropanol.
U.S. Pat. No. 5,722,720 (the ’720 patent) discloses a non-conventional technique for reducing particle size. In particular, the ’720 patent discloses a multistep process in which ondansetron hydrochloride dihydrate is first dried at elevated temperature and reduced or atmospheric pressure, and is then cooled to ambient temperature. The process requires the heating step to be performed until the ondansetron hydrochloride dihydrate is desolvated, and requires the cooling step to be performed until the ondansetron hydrochloride is rehydrated to form ondansetron hydrochloride dihydrate.
The ’720 patent process has several disadvantages. First, the ’720 patent process requires a prolonged time period (i.e., 16-24 hours) for the drying/desolvating step, plus an additional prolonged time period for the cooling/rehydrating step. Second, the ’720 patent process requires vigorous and carefully controlled drying conditions. For example, when the drying step is performed at 48-52° C., a reduced pressure of 100-200 torr is required. When the drying step is performed at ambient pressure, an elevated temperature of 1 00° C. is required.
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
Marcus Baumann , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK
Corresponding author email
Editor-in-Chief: J. Clayden
Beilstein J. Org. Chem. 2011, 7, 442–495.
A completely different strategy was used in the synthesis of the serotonin 5-HT3 receptor antagonist ondansetron (119, Zofran). In this synthesis a palladium-catalysed intramolecular Heck-reaction was used to build the tricyclic indole core in a short and concise sequence (Scheme 26) [35,36].
![[1860-5397-7-57-i26]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i26.png?max-width=550&background=FFFFFF)
Scheme 26: Palladium-mediated synthesis of ondansetron.
Alternatively, a direct Fischer indole synthesis between phenylmethyl hydrazine and a cyclic 1,3-dione derivative could be utilised to prepare the desired fully substituted tricyclic core of ondansetron (Scheme 27) [37].
![[1860-5397-7-57-i27]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i27.png?max-width=550&background=FFFFFF)
Scheme 27: Fischer indole synthesis of ondansetron.
- 35………..Godfrey, N.; Coates, I. H.; Bell, J. A.; Humber, D. C.; Ewan, G. B. Process for Preparing N-Heterocyclic Compounds. U.S. Patent 4,957,609, Sept 18, 1990.
- 36…………Iida, H.; Yuasa, Y.; Kibayashi, C. J. Org. Chem. 1980, 45, 2938–2942. doi:10.1021/jo01303a003
- Oxford, A. W.; Eldred, C. D.; Coates, I. H.; Bell, J. A.; Humber, D. C.; Ewan, G. B. Process for Preparing Tetrahydrocarbazolones. U.S. Patent 4,739,072, April 19, 1988.
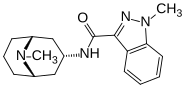
GRANISETRON
1-methyl-N-((1R,3r,5S)-9-methyl-9-azabicyclo[3.3.1]nonan-3-yl)-1H-indazole-3-carboxamide
107007-99-8 hydrochloride
109889-09-0 (free base)
109889-09-0 (free base)
AB-1001
ALM-101
BRL-43694
Inno-P08002
SP-01
SyB D-0701
SyB L-0701
SyB-0701
TRG
ALM-101
BRL-43694
Inno-P08002
SP-01
SyB D-0701
SyB L-0701
SyB-0701
TRG
gsk
Granisetron is a serotonin 5-HT3 receptor antagonist used as an antiemetic to treat nausea and vomiting following chemotherapy. Its main effect is to reduce the activity of the vagus nerve, which is a nerve that activates the vomiting center in the medulla oblongata. It does not have much effect on vomiting due to motion sickness. This drug does not have any effect on dopamine receptors or muscarinic receptors.
Granisetron hydrochloride is an anti-emetic drug, used for treatment or prophylaxis of emesis and post operative nausea and vomiting. Granisetron hydrochloride is marketed under the trade name Kytril as solution for injection as well as tablets. The chemical name of granisetron is N-(endo-9-methyl-9- azabicyclo[3.3.2]non-3-yl)-l-methylindazole-3-carboxamide and it is represented by the following structural formula :

Granisetron is usually administered as the hydrochloride salt for relieving the symptoms of vomiting and nausea in cancer patients. Recently the U.S. Food and Drug Administration (FDA) has accepted an investigational New Drug (IND) application for transdermal granisetron patch, Sancuso™, for the prevention of chemotherapy-induced nausea and vomiting (CINV). The Sancuso™ Phase I I Istudy is now underway in Europe and in the U.S. Typically, a non-oral form such as transdermal patch uses granisetron base as the active ingredient. The preparation of granisetron base is described in U.S. Patent No. 6,268,498 without referring to the solid state characteristics of granisetron. The preparation of granisetron base is further described in example 3 of U.S. Patent No. 7,071,209 (hereinafter the ’209 patent), having a melting point of 121-1220C. The ’209 patent is silent with regard to the solid state of granisetron base as well as to the solid state of the hydrochloride salt,
Granisetron was developed by chemists working at the British drug company Beecham around 1988 and is available as a generic. It is produced byRoche Laboratories under the trade name Kytril. The drug was approved in the United Kingdom in 1991 and in United States in 1994 by the FDA.
A granisetron transdermal patch with the trade name Sancuso was approved by the US FDA on September 12, 2008.[1] Sancuso is manufactured by ProStrakan, Inc., a pharmaceutical company headquartered in Bedminster, NJ, with global headquarters in Scotland.
Granisetron is metabolized slowly by the liver, giving it a longer than average half-life. One dose usually lasts 4 to 9 hours and is usually administered once or twice daily. This drug is removed from the body by the liver and kidneys.
Granisetron hydrochloride is a 5-HT3 antagonist that was launched in 1991 at Roche for the oral treatment of nausea. Preclinical studies demonstrate that, in binding to 5-HT3 receptors, granisetron blocks serotonin stimulation and subsequent vomiting after emetogenic stimuli such as cisplatin. In 2008, FDA approval of a transdermal patch was obtained by ProStrakan for the prophylaxis of chemotherapy-induced nausea/vomiting. Commercial launch took place the same year. This formulation has been filed for approval in the E.U. for the prevention of chemotherapy-induced nausea and vomiting. E.U. approval was obtained in 2012. In 2013, launch took place in United Kingdom. In 2011, Chugai Pharmaceutical received approval in Japan for the prevention of nausea and vomiting associated with antineoplastic agent administration and radiotherapy. Translational Research has developed an intranasal formulation that is in the preclinical phase of development. Acrux has also studied a proprietary metered-dose transdermal system, but progress reports on this formulation are not presently available. Currently marketed formulations include an oral solution, film-coated tablets, injections and sachets. BioDelivery Sciences is developing a formulation of granisetron hydrochloride using its film technology (BioErodable MucoAdhesive) BEMA technology. Almac is developing the compound in phase I clinical studies for the prevention of chemotherapy-induced nausea/vomiting.
It may be used for chemotherapy-induced nausea and vomiting and appears to work about the same as ondansetron.[2]
A number of medications including granisetron appear to be effective in controlling post-operative nausea and vomiting (PONV).[3] It is unclear if it is better than or worse than other agents like droperidol, metoclopramide, ondansetron or cyclizine.[3]
Its efficacy has also been questioned with a research Dr. Yoshitaka Fujii having 12 published papers on this topic in Canadian Journal of Anesthesia retracted. A further five papers in the same journal on the same drug by Dr Fujii are considered indeterminate.
- Is a possible therapy for nausea and vomiting due to acute or chronic medical illness or acute gastroenteritis
- Treatment of cyclic vomiting syndrome although there are no formal trials to confirm efficacy.
Granisetron is a well-tolerated drug with few side effects. Headache, dizziness, and constipation are the most commonly reported side effects associated with its use. There have been no significant drug interactions reported with this drug’s use. It is broken down by the liver‘s cytochrome P450 system and it has little effect on the metabolism of other drugs broken down by this system.
APF530
A New Drug Application (NDA) for APF530, a sustained-delivery form of Granisetron, was accepted in October 2012.[4] APF530 will be targeted as anantiemetic, towards patients undergoing radiation therapy and chemotherapy. APF530 contains the 5-HT3 antagonist, granisetron, formulated in the Company’s proprietary Biochronomer™ drug delivery system, which allows therapeutic drug levels to be maintained for five days with a single subcutaneous injection.
Originally developed at GlaxoSmithKline, granisetron hydrochloride was divested in September 2000 giving Roche global rights to the drug. Currently, granisetron is being distributed by Roche in France, Italy, South Africa, the U.K. and the U.S. and in Japan by Roche’s subsidiary Chugai. In 2007, a license agreement was signed between LG Life Sciences and ProStrakan in Korea. In 2008, the product was licensed to JapanBridge by ProStrakan for development and marketing in Asia for the prophylaxis of chemotherapy-induced nausea and vomiting. An additional license agreement was made in 2008 granting Paladin rights to granisetron transdermal patches for the treatment of nausea. In 2010, granisetron hydrochloride extended-release transdermal patches were licensed to Kyowa Hakko Kirin by Solasia Pharma in Taiwan, Hong Kong, Singapore and Malaysia for the prevention of chemotherapy-induced nausea and vomiting. Solasia retains full rights in Japan and China.
- Kytril Web site
- Sancuso Web siteKYTRIL Tablets and KYTRIL Oral Solution contain granisetron hydrochloride, an antinauseant and antiemetic agent. Chemically it is endo-N-(9-methyl-9-azabicyclo [3.3.1] non-3-yl)-1-methyl-1H-indazole-3-carboxamide hydrochloride with a molecular weight of 348.9 (312.4 free base). Its empirical formula is C18H24N4O•HCl, while its chemical structure is:
 granisetron hydrochlorideGranisetron hydrochloride is a white to off-white solid that is readily soluble in water and normal saline at 20°C.
granisetron hydrochlorideGranisetron hydrochloride is a white to off-white solid that is readily soluble in water and normal saline at 20°C.
- PRNewswire. FDA Approves Sancuso, the First and Only Patch for Preventing Nausea and Vomiting in Cancer Patients Undergoing Chemotherapy. September 12, 2008.
- Billio, A; Morello, E; Clarke, MJ (2010 Jan 20). “Serotonin receptor antagonists for highly emetogenic chemotherapy in adults.”. The Cochrane database of systematic reviews (1): CD006272.PMID 20091591.
- Carlisle, JB; Stevenson, CA (2006 Jul 19). “Drugs for preventing postoperative nausea and vomiting.”. The Cochrane database of systematic reviews (3): CD004125. PMID 16856030.
- Drugs.com A.P. Pharma Announces PDUFA Action Date for APF530 New Drug Application Resubmission. October 16, 2012.
- Katzung, Bertram G. Basic and Clinical Pharmacology, 9th ed. (2004). ISBN 0-07-141092-9
- Aapro, M. (2004). “Granisetron: an update on its clinical use in the management of nausea and vomiting”. The oncologist 9 (6): 673–686. doi:10.1634/theoncologist.9-6-673. ISSN 1083-7159. PMID 15561811. edit
| EP0200444A2 * | Apr 21, 1986 | Nov 5, 1986 | Beecham Group Plc | Azabicyclononyl-indazole-carboxamide having 5-HT antagonist activity |
| EP1484321A1 * | May 27, 2004 | Dec 8, 2004 | Chemagis Ltd. | Process for preparing 1-methylindazole-3-carboxylic acid |
| WO1995023799A1 * | Feb 28, 1995 | Sep 8, 1995 | Victor Witold Jacewicz | Process for the preparation of an indazole-3-carboxamide derivative |
| WO1997030049A1 * | Feb 11, 1997 | Aug 21, 1997 | Victor Witold Jacewicz | Process for the preparation of granisetron |
| EP0200444A2 * | Apr 21, 1986 | Nov 5, 1986 | Beecham Group Plc | Azabicyclononyl-indazole-carboxamide having 5-HT antagonist activity |
| ES2129349A * | Title not available |
| 1 | * | BERMUDEZ J. ET AL.: ‘5-Hydroxytryptamine (5-HT3) receptor antagonists. 1. Indazole and indolizine-3-carboxylic acid derivatives‘ J. MED. CHEM. vol. 33, no. 7, 1990, pages 1924 – 1929 |
| PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| WO2008117282A1 * | Mar 24, 2008 | Oct 2, 2008 | Itai Adin | Polymorph of granisetron base and production process therefor |
| EP2323654A1 * | Aug 18, 2009 | May 25, 2011 | ScinoPharm Taiwan, Ltd. | Polymorphic form of granisetron hydrochloride and methods of making the same |
| WO2003080606A1 * | Mar 21, 2003 | Oct 2, 2003 | Barjoan Pere Dalmases | Process for preparing a pharmaceutically active compound (granisetron) |
| WO2007054784A1 * | Nov 8, 2006 | May 18, 2007 | Shanmuga Sundaram Bharan Kumar | An improved process for the preparation of granisetron hydrochloride |
| WO2007088557A1 * | Jan 18, 2007 | Aug 9, 2007 | Prasad Ramanadham Jyothi | Process for highly pure crystalline granisetron base |
| ES2124162A1 * | Title not available |
| WO2007007886A1 * | Jul 10, 2006 | Jan 18, 2007 | Tanabe Seiyaku Co | An oxime derivative and preparations thereof |
| WO2007088557A1 * | Jan 18, 2007 | Aug 9, 2007 | Prasad Ramanadham Jyothi | Process for highly pure crystalline granisetron base |
| WO2008117282A1 * | Mar 24, 2008 | Oct 2, 2008 | Itai Adin | Polymorph of granisetron base and production process therefor |
| WO2008151677A1 | Dec 20, 2007 | Dec 18, 2008 | Inke Sa | Polymorphic form of granisetron base, methods for obtaining it and formulation containing it |
| EP2164848A1 * | Dec 20, 2007 | Mar 24, 2010 | Inke, S.A. | Polymorphic form of granisetron base, methods for obtaining it and formulation containing it |
| EP2323654A1 * | Aug 18, 2009 | May 25, 2011 | ScinoPharm Taiwan, Ltd. | Polymorphic form of granisetron hydrochloride and methods of making the same |
| US8193217 * | Aug 18, 2009 | Jun 5, 2012 | Scinopharm Taiwan Ltd. | Polymorphic form of granisetron hydrochloride and methods of making the same |
| US20100048613 * | Aug 18, 2009 | Feb 25, 2010 | Scinopharm Taiwan Ltd. | Polymorphic form of granisetron hydrochloride and methods of making the same |
| WO2007088557A1 * | Jan 18, 2007 | Aug 9, 2007 | Prasad Ramanadham Jyothi | Process for highly pure crystalline granisetron base |
| WO2008117282A1 * | Mar 24, 2008 | Oct 2, 2008 | Itai Adin | Polymorph of granisetron base and production process therefor |
| WO2008151677A1 | Dec 20, 2007 | Dec 18, 2008 | Inke Sa | Polymorphic form of granisetron base, methods for obtaining it and formulation containing it |
| EP2164848A1 * | Dec 20, 2007 | Mar 24, 2010 | Inke, S.A. | Polymorphic form of granisetron base, methods for obtaining it and formulation containing it |
| EP2323654A1 * | Aug 18, 2009 | May 25, 2011 | ScinoPharm Taiwan, Ltd. | Polymorphic form of granisetron hydrochloride and methods of making the same |
Drugs Fut 1989, 14(9): 875
5-Hydroxytryptamine (5-HT3) receptor antagonists. 1. Indazole and indolizine-3-carboxylic acid derivatives
J Med Chem 1990, 33(7): 1924
J Med Chem 1990, 33(7): 1924
WO 2007054784
US 4886808
US 5034398
US 5034398
IN 200901669
……………………………………………………………………………………………………………………………………
Granisetron hydrochloride of formula (I). More particularly this invention relates to the preparation of Granisetron hydrochloride using methyl isobutyl ketone (MIBK) as a single solvent in presence of an organic base such as triethylamine.

(I)
Granisetron hydrochloride which is chemically known as endo-l-methyl-N-(9- methyl-9-azabicyclo[3.3.1]non-3-yl)-lH-indazole-3-carboxamide monohydrochloride is a 5-HT (5 -hydroxy triptamine) antagonist, and has the following structural formula:

(I)
Granisetron hydrochloride is useful as an anti-emetic and marketed as Kytril by Roche.
EP-A-0200444 provides certain 5-HT (5-hydroxytryptamine) antagonists, which are described as possessing a number of therapeutic utilities, inter alia, the prevention of vomiting following the administration of cytotoxic agents. The compound described in Example 6 is endo-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)- l-methylindazole-3-carboxamide, and this compound has been assigned the INN Granisetron. EP-A-0200444 also discloses that Granisetron can be prepared by reacting l-methylindazole-3-carboxylic acid chloride with endo-3-amino-9-methyl-9-azabicyclo [3.3.1] nonane.
EP 748321 claims a process for preparing Granisetron or a pharmaceutical acceptable salt thereof. The process comprises the condensation of compound of formula (3) and (4) followed by de-protecting the intermediate compound of structure (2) to get the granisetron or optionally forming a pharmaceutically accepted salt of Granisetron. The scheme is presented below in which Q is a leaving group displaceable by a secondary amine wherein R may be represented as benzyl, benzyl substituted with one or more chloro, alkyl or alkoxy group, t-butyl, allyl or a t-butyldimethylsilylgroup.

GRANISETRON
US Pat. No. 6,268,498 discloses an alternative process for preparing
Granisetron, by cyclisation of a previously methylated compound of formula (C), which is shown below. It should be noted that the methylation prior to cyclisation is carried out with sodium hydride and methyl iodide as disclosed in example 1 (b) of said patent. However, the cyclisation conditions applied to that compound of formula (C) may facilitate demethylation of the indazole of the Granisetron so obtained. Thus, for example, in examples 2 and 3 of said patent described the cyclisation reaction, but although in example 2 the reaction leads to Granisetron, in example 3, when the reaction time is increased under the same conditions, quantitatively demethylated Granisetron is provided. The reaction time therefore has a consideration influence on the yield values in the second step of the process that is in the cyclisation, since the Granisetron provided by this process contains as an impurity significant amounts of demethylated Granisetron, which will have to be re-methylated in an additional step.

(C) ES 2,124,162 patent discloses a procedure for the preparation of Granisetron or its pharmaceutically acceptable salts consisting of reaction of l-methylindazole-3- carboxamide of formula (A) with 9-methyl-9-azabicyclononane of formula (B) (L = halogen, OMs, OTs; halogen = esp. Cl, Br; Ms = SO2Me; Ts = SO2C6H4Me4). Thus, l-methylindazole-3-carboxamide in tetrahydrofuran (THF) containing tetramethylethylenediamine is treated with BuLi in hexane followed by addition of endo-3-(mesyloxy)-9-methyl-9-azabicyclo [3.3.1] nonane hydrochloride (L = OMs) to give the title compound i.e. Granisetron The scheme is represented below:

(A) (B) GRANISETRON
As discussed above none of the prior art references disclosed or claimed the use of methyl isobutyl ketone (MIBK) as a single solvent in presence of an organic Λ
4
base such as triethylamine for the preparation of compound of formula (I), hence we focused our research to develop an improved and efficient process for the preparation of the compound of formula (I) in substantial good yield and high purity.
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
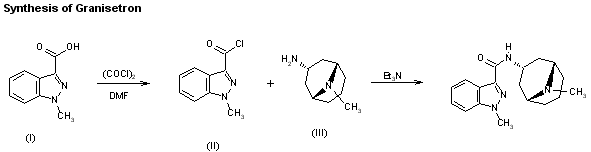
................................................................................................
The synthesis of granisetron has been reported: The reaction of 1-methylindazole-3-carboxylic acid (I) with oxalyl chloride and DMF in dichloromethane gives 1-methylindazole-3-carbonyl chloride (II), which is then condensed with endo-9-methyl-9-azabicyclo[3.3.1]nonan-3-amine (III) by means of triethylamine in dichloromethane.
AU 8656579; EP 0200444; EP 0223385; EP 0498466; ES 8707948; JP 1986275276; JP 1993194508; US 4886808; US 5034398
................................................................................................

Example (1) Preparation of Granisetron hydrochloride
10OmL of methyl isobutyl ketone (MIBK), 8.6g of triethylamine and 1Og of 1- methyl indazole-3-carboxylic acid were placed in a 25OmL RBF. The reaction mass was stirred and treated with 7.4g of ethyl chloro formate at O0C to (-) 50C to get a mixed anhydride and then condensed with 8.75g of endo-9-methyl-9-azabicycolo [3.3.1] nonan-3-amine and stirred the reaction mass till the completion of reaction. To the reaction mixture 100 mL of water was added, organic layer separated and distilled to 8 volumes of MIBK, cooled the reaction mass and treated with 10.3g of IPA/HC1 (~ 20%) to get 1Og of Granisetron hydrochloride. Example (2) Preparation of Granisetron base
75OmL of methyl isobutyl ketone, 4Og of triethylamine and 5Og of 1-methyl indazole-3-carboxylic acid were placed in a 2L RBF. The reaction mass was stirred and treated with 34g of ethyl chloro formate at 20°C to 25°C to get a mixed anhydride and then condensed with 48g of endo-9-methyl-9-azabicycolo [3.3.1] nonan-3-amine and stirred the reaction mass till the completion of reaction. To the reaction mixture 500 mL of water was added and the organic layer separated, washed with 5% sodium carbonate
(50OmL) solution and distilled the organic layer to obtain Granisetron freebase with HPLC purity 99.91%.
Example (2 – a ) Preparation of Granisetron hydrochloride
1Og of Granisetron freebase, 10OmL of methanol were placed in 250 mL RBF and heated the reaction mass to 400C to 550C to get a clear solution. The clear solution was filtered and treated with 3.5g of concentrated hydrochloric acid (36%) and diluted the reaction mass with 20OmL of MIBK, heated the reaction mass to 600C to 65°C and distilled the reaction mass up to 10 to 12 Volumes. The reaction mass was cooled and isolated 1Og of Granisetron hydrochloride with HPLC purity 99.91%.
………………………………………………………………………………………………………………………….
Example 3 Preparation of endo-N-(9-methyl-9-azabicyclo[3.3. l]non-3-yl)-indazole-3- carboxamide.
A solution of 2-(N-methylbenzylidenehydrazino)-α-oxophenyl-[endo-N-(9- methyl-9-azabicyclo[3.3.1]non-3-yl)] carboxamide(0.536 g) in methanol (8 ml) was treated with 2N hydrochloric acid (0.4 ml) at room temperature. A rapid colour change from orange to green was observed. The solution was stirred for 24 hours then evaporated to the give the crude endo-N-(9-methyl-9-azabicyclo[3.3.1] non-3-yl)-indazole-3-carboxamide (0.630g).
Example 4 Preparation of endo-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)-l-methylindazole- 3-cjχboxamide (granisetron).
Sodium hydride (72 mg, 60% dispersion in oil) was added to a solution of endo- N-(9-methyl-9-azabicyclo[3.3. l]non-3-yl)-indazole-3-carboxamide (0.130g) in dry tetrahydrofuran (3.0 ml) under nitrogen at -50°C. The resultant solution was warmed to room temperature over 20 minutes then cooled to -40°C and treated with methyl iodide (0.015 ml). After 3 hours at room temperature HPLC analysis showed complete conversion to endo-N-(9-methyl-9-azabicyclo[3.3.1] non-3-yl)- l-methy-indazole-3-carboxamide. Water (10 ml) was added and the mixture extracted with ethyl acetate (2x 20 ml). The extracts were dried (MgSO4) ^ evaporated to give endo-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)-l- methylindazole-3-carboxamide 50 mg (41%). MS 313 (M+H)+.
''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
5 ALOSETRON

ALOSETRON
5-methyl-2-[(4-methyl-1H-imidazol-5-yl)methyl]-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indol-1-one
132414-02-9, Hydrochloride
122852-42-0 (free base)
122852-42-0 (free base)
122852-69-1 CHECK…..
GR-68755C
- Alosetron HCl
- Alosetron hydrochloride
- GR 68755c
- HSDB 7055
- Lotronex
- UNII-2F5R1A46YW
GSK
LAUNCHED 2002
| United States PATENT | US5360800 | APPROVED1993-01-13 | EXPIRY 2013-01-13 |
Alosetron is a 5-HT3 antagonist used only for the management of severe diarrhoea-predominant irritable bowel syndrome (IBS) in women. Alosetron has an antagonist action on the 5-HT3 receptors and thus may modulate serotonin-sensitive gastrointestinal (GI) processes. Alosetron was voluntarily withdrawn from the US market in November 2000 by the manufacturer due to numerous reports of severe adverse effects including ischemic colitis, severely obstructed or ruptured bowel, and death. In June 2002, the FDA approved a supplemental new drug application allowing the remarketing of the drug under restricted conditions of use.
Alosetron hydrochloride (initial brand name: Lotronex; originator: GSK) is a 5-HT3 antagonist used for the management of severe diarrhea-predominant irritable bowel syndrome (IBS) in women only. It is currently marketed by Prometheus Laboratories Inc. (San Diego), also under the trade name Lotronex. Alosetron was withdrawn from the market in 2000 owing to the occurrence of serious life-threatening gastrointestinal adverse effects, but was reintroduced in 2002 with availability and use restricted.
Alosetron hydrochloride is a potent and selective 5-HT3 antagonist marketed by GlaxoSmithKline for the oral treatment of irritable bowel syndrome (IBS) in female patients whose predominant bowel symptom is diarrhea. It is currently marketed in a tablet formulation. In 2000, the drug was withdrawn from several markets based on adverse reactions, however, was reintroduced on the U.S. market in 2002 following a recommendation of a joint FDA advisory panel comprising members of the Gastrointestinal Drugs Advisory Committee and the Drug Safety and Risk Management Subcommittee of the Pharmaceutical Science Committee which stipulated reintroduction of the drug in conjunction with a risk management plan.

Alosetron was originally approved by the U.S. Food and Drug Administration (FDA) on February 9, 2000,[1] after a seven month review.[2] At the time of the initial approval U.S. Food and Drug Administration (FDA) reviewers found that alosetron improved symptoms in 10% to 20% of patients.[3]
Shipment to pharmacies started in March, 2000. On July 17, a health professional filed a report with the FDA on the death of a 50-year-old woman who suffered mesenteric ischemia. The report identified alosetron as the “primary suspect” in the death.[4]
Alosetron was withdrawn from the market voluntarily by GlaxoWellcome on November 28, 2000 owing to the occurrence of serious life-threateninggastrointestinal adverse effects, including 5 deaths and additional bowel surgeries.[2] The FDA said it had reports of 49 cases of ischemic colitis and 21 cases of “severe constipation” and that ten of the 70 patients underwent surgeries and 34 others were examined at hospitals and released without surgery. Through November 17, 2000, pharmacists had filled 474,115 prescriptions for Alosetron.[2] Severe adverse events continued to be reported, with a final total of 84 instances of ischaemic colitis, 113 of severe constipation, 143 admissions to hospital, and 7 deaths.[5]
Patient advocacy groups, most notably the Lotronex Action Group and the International Foundation for Functional Gastrointestinal Disorders (IFFGD) lobbied for the drug’s return. Public Citizen Health Research Group, another patient advocacy group, opposed the reintroduction.[6][7]
On June 7, 2002, the FDA announced the approval of a supplemental New Drug Application (sNDA) that allows restricted marketing of Lotronex (alosetron hydrochloride), to treat only women with severe diarrhea-predominant irritable bowel syndrome (IBS).[8] It was the first drug ever returned to the U.S. market after withdrawal for safety concerns.[9][10]
It is not known whether alosetron has been filed for registration in the EU.
GSK sold Lotronex to the Californian corporation Prometheus in late 2007.[11]
Alosetron hydrochloride works through antagonism of the serotonin 5-HT3 receptor, distributed extensively on visceral neurons in the human gastrointestinal tract, as well as other peripheral and central locations. Activation of 5-HT3 channels results in neuronal depolarization and affects the regulation of visceral pain, colonic transit and gastrointestinal secretions. Alosetron inhibits activation of non-selective cation channels and modulates the enteric nervous system. In previous clinical trials, the drug increased colonic transit time without affecting orocecal transit time, increased basal jejunal water and sodium absorption and significantly increased colonic compliance. In 2007, the compound was licensed to Prometheus Laboratories by GlaxoSmithKline in the U.S. for the treatment of IBS in patients whose predominant bowel symptom is diarrhea.
Criticism of the FDA
In 2001, the editor of the renowned medical journal The Lancet, Richard Horton, criticized the FDA’s handling of alosetron in an unusually sharp language.[12] Horton argued that the treatment of a non-fatal condition did not justify the use of a drug with potentially lethal side effects, and that the FDA should have revoked the approval for alosetron sooner when postmarketing surveillance revealed that many patients had suffered constipation necessitating surgical intervention and ischaemic colitis. He asserted that FDA officials were improperly motivated to maintain and reinstate the approval for alosetron because of the extent to which the FDA’s Center for Drug Evaluation and Research is funded by user fees paid by pharmaceutical manufacturers, and that the reinstatement of alosetron was negotiated in confidential meetings with representatives ofGlaxoSmithKline.
An article published in the British Medical Journal (BMJ) noted: “By allowing the marketing of alosetron, a drug that poses a serious and significant public health concern according to its own terms, the FDA failed in its mission.”[13] Others have argued that the approval process of Lotronex was an example of regulatory capture.[7]
Alosetron has an antagonist action on the 5-HT3 receptors of the enteric nervous system of the gastrointestinal tract. While being a 5-HT3 antagonist like ondansetron, it is not classified or approved as an antiemetic. Since stimulation of 5-HT3 receptors is positively correlated with gastrointestinal motility, alosetron’s 5-HT3 antagonism slows the movement of fecal matter through the large intestine, increasing the extent to which water is absorbed, and decreasing the moisture and volume of the remaining waste products.[14]
- U.S. Food and Drug Administration. “Drug Details”. Retrieved 11 December 2012.
- Willman, David (29 November 2000). “Drug Lotronex Pulled Over Safety Fears”. The Los Angeles Times. Retrieved 11 December 2012.
- Willman, David (20 December 2000). “Officer Foresaw Deadly Effects”. The Los Angeles Times. Retrieved 11 December 2012.
- Willman, David (2 November 2000). “FDA Minimized Issue of Lotronex’s Safety”. The Los Angeles Times. Retrieved 11 December 2012.
- CENTER FOR DRUG EVALUATION AND RESEARCH (23 April 2002). “GASTROINTESTINAL DRUGS ADVISORY COMMITTEE AND DRUG SAFETY AND RISK MANAGEMENT SUBCOMMITTEE OF THE ADVISORY COMMITTEE FOR PHARMACEUTICAL SCIENCE”. U.S. Food and Drug Administration. Retrieved 11 December 2012.
- Grady, Denise (23 April 2002). “Appeals Prompt U.S. Agency to Consider Allowing Sales of Diarrhea Drug Linked to Deaths”. The New York Times. Retrieved 11 December 2012.
- Moynihan, Ray (14 September 2002). “Alosetron: a case study in regulatory capture, or a victory for patients’ rights?”. The British Medical Journal 325 (7364): 592–595.PMC 1124108. PMID 12228140. Retrieved 11 December 2012.
- U.S. Food and Drug Administration. “Lotronex (alosetron hydrochloride) Information”. U.S. Food and Drug Administration. Retrieved 11 December 2012.
- Pollack, A (2006-03-09). “F.D.A. Panel Recommends M.S. Drug Despite Lethal Risk”. The New York Times. Retrieved 2008-03-13.
- Grady, Denise (8 June 2002). “U.S. Lets Drug Tied to Deaths Back on Market”. The New York Times. Retrieved 11 December 2012.
- Prometheus Laboratories Inc. Press Release of 7 November 2007. Retrieved on 27 August 2008.
- Horton, R. (2001). “Lotronex and the FDA: a fatal erosion of integrity”. The Lancet 357 (9268): 1544–1545. doi:10.1016/S0140-6736(00)04776-0. edit
- Lièvre, Michel (14 September 2002). “Alosetron for irritable bowel syndrome”. The British Medical Journal 325 (7364): 555–556. PMC 1124090. PMID 12228116. Retrieved 11 December 2012.
- “HIGHLIGHTS OF PRESCRIBING INFORMATION”. Prometheus Laboratories Inc. April 2008. Retrieved 11 December 2012.
- Camilleri, M.; Northcutt A.R., Kong S., Dukes G.E., McSorley D., Mangel A.W. (25 March 2000). “Efficacy and safety of alosetron in women with irritable bowel syndrome: a randomised, placebo-controlled trial.”. The Lancet 355 (1035): 1035–40. doi:10.1016/S0140-6736(00)02033-X. PMID 10744088.
- Barbehenn, Elizabeth; Peter Lurie, Sidney M. Wolfe (9 December 2000). “Alosetron for irritable bowel syndrome”. The Lancet 356 (9246): 2009. doi:10.1016/S0140-6736(05)72978-0. Retrieved 11 December 2012.
IMPORTANT REF
Drugs Fut 1992, 17(8): 660
US 2012178937
JP 2012140415
WO 2010121038
US 200815392
WO 2006119329
JP 2005225844
WO 1999017755
WO 2001087305
WO 2001045685
US 5229407
US 5008272
9-22-2010
|
Pyrimidine derivatives
| |
5-7-2010
|
INDOLONE MODULATORS OF 5-HT3 RECEPTOR
| |
10-3-2003
|
Method for treating functional dyspepsia
| |
7-16-2003
|
Methods for treating irritable bowel syndrome
| |
8-7-2002
|
Methods for treating irritable bowel syndrome
| |
9-5-2001
|
Medicaments for the treatment of non-constipated female irritable bowel syndrome
| |
12-22-2000
|
RECEPTOR AGONISTS AND ANTAGONISTS COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION
| |
8-25-2000
|
USE OF 5-HT3 RECEPTOR ANTAGONISTS USE OF 5-HT3 RECEPTOR ANTAGONISTS FOR TREATING MUSCULOESKELETAL DISEASES
| |
8-25-2000
|
SYSTEMIC USE OF 5-HT3 RECEPTOR ANTAGONISTS AGAINST RHEUMATIC INFLAMMATORY PROCESSES
| |
7-20-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
5-32-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS
| |
12-30-1999
|
METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS
| |
12-17-1999
|
MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT>3< RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS
| |
6-25-1999
|
ORAL DELIVERY FORMULATION
| |
4-16-1999
|
MEDICAMENTS MEDICAMENTS
| |
8-19-1998
|
DHA-pharmaceutical agent conjugates of taxanes
| |
11-28-1997
|
DHA-PHARMACEUTICAL AGENT CONJUGATES DHA-PHARMACEUTICAL AGENT CONJUGATES
| |
3-28-1997
|
5-HT3 RECEPTOR ANTAGONISTS FOR DYSKINESIA
| |
11-2-1994
|
Tetrahydro-1H-pyrido[4,3-b]indol-1-one derivatives
| |
8-31-1994
|
Controlled release device
|
9-22-2010
|
Pyrimidine derivatives
| |
5-7-2010
|
INDOLONE MODULATORS OF 5-HT3 RECEPTOR
| |
10-3-2003
|
Method for treating functional dyspepsia
| |
7-16-2003
|
Methods for treating irritable bowel syndrome
| |
8-7-2002
|
Methods for treating irritable bowel syndrome
| |
9-5-2001
|
Medicaments for the treatment of non-constipated female irritable bowel syndrome
| |
12-22-2000
|
RECEPTOR AGONISTS AND ANTAGONISTS COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION
| |
8-25-2000
|
USE OF 5-HT3 RECEPTOR ANTAGONISTS USE OF 5-HT3 RECEPTOR ANTAGONISTS FOR TREATING MUSCULOESKELETAL DISEASES
| |
8-25-2000
|
SYSTEMIC USE OF 5-HT3 RECEPTOR ANTAGONISTS AGAINST RHEUMATIC INFLAMMATORY PROCESSES
| |
7-20-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
The Alosetron hydrochloride is a potent and selective antagonist of the serotonin 5-HT3 receptor type. Chemically, Alosetron is designated as 2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-1H-imidazol-4-yl)methyl]-1H-pyrido[4,3-b]indol-1-one, monohydrochloride. This is marketed in United States under trade name of LOTRONEX®
U.S. Pat. No. 5,360,800 discloses a preparation of Alosetron by condensing 2,3,4,5-tetrahydro-5-methyl-1H-pyrido[4,3-b]indol-1-one with 4-chloromethyl-5-methylimidazole in presence of strong base such as sodium hydride. The sodium hydride was corrosive and highly flammable. This type of reaction required extra care, special type of equipments and it is commercially not feasible. This process also provides low yield.
U.S. Pat. No. 6,175,014 patent describes a process for the process Alosetron by reacting of 2,3,4,5-tetrahydro-5-methyl-1H-pyrido[4,3-b]indol-1-one of formula (II) with 4-hydroxymethyl-5-methylimidazole of formula (IIIa) or its salt in presence of mineral acid like hydrochloric acid or sulfonic acids such as p-toluene sulfonic acid or methane sulfonic acid. The process requires purification to get pure Alosetron.

Hence there is a need for a simple and commercially viable process for the preparation of Alosetron which avoids hazardous base such as sodium hydride.
The present inventors identified a new process for the preparation of Alosetron by reaction of 2,3,4,5-tetrahydro-5-methyl-1H-pyrido[4,3-b]indol-1-one of formula (II) with 4-hydroxymethyl-5-methylimidazole of formula (III) or its protected derivative. This process is simple to carryout for large scale preparation and industrially viable
medical use for compounds which act as antagonists of 5-hydroxytryptamine (5-HT) at 5-HT3 receptors.
5-HT3 receptor antagonists may be identified by methods well known in the art, for example by their ability to inhibit 3-(5-methyl-1H-imidazole-4-yl)-1-[1-[3H]- methyl-1 H-indol-3-yl]-1-propanone binding in rat entorhinal cortex homogenates (following the general procedure described by G Kilpatrick et al, Nature, 1987, 330, 746-748), and/or by their effect on the 5-HT-induced Bezold-Jarisch (B-J) reflex in the cat (following the general method described by A Butler et al, Br. J. Pharmacol., 94, 397-412 (1988)).
A number of different 5-HT3 receptor antagonists have been disclosed, for example those of group A: indisetron, Ro-93777, YM-114, granisetron, talipexole, azasetron, tropisetron, mirtazapine, ramosetron, ondansetron, lerisetron, alosetron, N-3389, zacopride, cilansetron, E-3620, lintopride, KAE- 393, itasetron, mosapride and dolasetron.
In UK Patent No. 2209335, incorporated herein by reference, there is disclosed, inter alia, the compound 2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-1H-imidazol-4- yl)methyl]-1H-pyrido[4,3-b]indol-1-one, now known as alosetron, which may be represented by the formula (I):

and pharmaceutically acceptable salts, solvates and pharmaceutically acceptable equivalents thereof, in particular its hydrochloride salt.
…………………………………………………………………………………………………………

EXAMPLE 1 Process for the Preparation of Alosetron
To a mixture of acetic acid and dimethylformamide, 3N-BOC-(-hydroxymethyl-5-methyl imidazole (95.4 g), 2,3,4,5-tetrahydro-5-methyl-1H-pyrido[4,3-b]indol-1-one (50 g), trifluoroacetic acid were added and heated to 100-115° C. After completion of the reaction, the reaction mass was cooled to room temperature. To the reaction mass, carbon was added, stirred and filtered though hyflo bed. The bed was washed with dimethylformamide. The filtrate was distilled under vacuum. To the residue, water was added and washed the reaction mass with toluene followed by isopropyl ether. The pH of the reaction mass was adjusted to 6.8-7 using potassium carbonate solution, stirred, cooled and the obtained solid was dried.
EXAMPLE 2 Process for the Preparation of Alosetron
To trifluoroacetic acid, 3N-BOC-4-hydroxymethyl-5-methylimidazole (95.4 g), dimethylformamide (480 mL) and 2,3,4,5-tetrahydro-5-methyl-1H-pyrido[4,3-b]indol-1-one (58 g) were added and heated to 100-115° C. After completion of the reaction, the reaction mass was cooled to room temperature. To the reaction mass, carbon was added and filtered though hyflo bed. The bed was washed with dimethylformamide and the filtrate was distilled under vacuum. To the residue, water was added and washed the aqueous layer with toluene followed by isopropyl ether. The pH of the reaction mass was adjusted to 6.8-7 using potassium carbonate solution, cooled and the obtained solid was dried.
| TABLE 1 | ||||
| Solvent System | Reaction time | Yield | ||
| TFA & DMF | 6-7 hours | 65% | ||
| acetic acid alone | 20 hours | 17% | ||
| acetic acid & DMF | No reaction | |||
The above table clearly indicates that the use trifluoroacetic acid (TFA) enhances the reaction progress and also increases the yield of the product.
EXAMPLE 3 Purification of Alosetron
To acetic acid, crude Alosetron containing >0.2% of compound of formula (IV) was added and heated to 60-65° C., the reaction mass maintained at same temperature and cooled to 40-45° C. To the reaction mass acetone was added and refluxed. The reaction mass was cooled to 0-5° C., the solid obtained was filtered, washed with acetone (slurry) and dried to yield pure Alosetron having less than 0.02% of Impurity of formula (IV) by HPLC. Yield: 53-50 g
Reference Example 1 Preparation of Alosetron Hydrochloride
To a methanol (50 mL), Alosetron (10 g) and of IPA.HCl (8.5 mL) were added and heated to 40-45° C. The reaction mass was cooled, stirred and filtered and washed with methanol. The reaction mass was dissolved in methanol, treated with carbon, filtered and washed with methanol. The reaction mass was distilled and isopropyl ether was added to the residue and stirred at room temperature. The reaction mass was cooled, stirred. The solid obtained was filtered and washed with chilled methanol and dried.
Yield: 7.8 g Reference Example-2 Process for the preparation of 3N-BOC-(4-hydroxymethyl)-5-methylimidazole
4-Hydroxymethyl-5-methylimidazole (100 g) was dissolved in water, to the solution was added sodium carbonate (107 g) and stirred. To the reaction mass acetonitrile (400 mL) was added and cooled to 10-15° C. followed by addition of solution of DIBOC (di-tert-butyl dicarbonate) in acetonitrile. After completion of the reaction, water was added to the reaction mass and filtered. The filtrate was washed with 1:1 acetonitrile and water and than washed with hexane. The mass was extracted with toluene and the organic layer was washed with water followed by brine. The organic layer was distilled under vacuum to get oily mass of the title compound.
……………………………………..

TROPISETRON
89565-68-4
105826-92-4 (HCl)
105826-92-4 (HCl)
ICF 205-930, Navoban™
Molecular Formula: C17H20N2O2 Molecular Weight: 284.3529
Novartis (Originator)
Tropisetron (INN) is a serotonin 5-HT3 receptor antagonist used mainly as an antiemetic to treat nausea and vomiting following chemotherapy, although it has been used experimentally as an analgesic in cases of fibromyalgia.[1] The drug is available in a 5 mg oral preparation or in 2 mg intravenous form. It is marketed by Novartis in Europe, Australia, New Zealand, Japan, South Korea and the Philippines as Navoban, but is not available in the U.S. It is also available from Novell Pharmaceutical Laboratories and marketed in several Asian countries as Setrovel
Tropisetron is a 5-hydroxytryptamine receptor 3 (5-HT3) antagonist that was launched in 1992 by Novartis for the oral and injection treatment of chemotherapy-induced emesis. The drug has also been approved for the prophylaxis and treatment of post-operative nausea and vomiting, and is available in capsule and ampule formulations. In terms of clinical development, phase III trials were being carried out by the National Institute of Mental Health (NIHM) as an adjunct to risperidone therapy for the treatment of schizophrenia, but no recent development has been reported. Clinical trials for the treatment of fibromyalgia have also been conducted by Novartis, although recent progress reports on this indication have not been made available.
5-HT3 receptors are excitatory ligand-gated cation channel receptors that can be found in the presynaptic vagal afferents, the area postrema and the gastrointestinal tract. Stimulation of these receptors seems to be important in the emetic response and the gag reflex. It has also been shown that tropisetron shows agonistic effects on the 7 nicotinic acetylcholine receptor. This receptor is associated with auditory sensory gating, a neural mechanism believed to have important roles in information processing and cognition, both diminished in people with schizophrenia.
Tropisetron acts as both a selective 5-HT3 receptor antagonist and α7-nicotinic receptor agonist.[2][3]

Tropisetron is a well-tolerated drug with few side effects. Headache, constipation, and dizziness are the most commonly reported side effects associated with its use. Hypotension, transient liver enzyme elevation, immune hypersensitivity syndromes and extrapyramidal side effects have also been associated with its use on at least one occasion.There have been no significant drug interactions reported with this drug’s use. It is broken down by the hepatic cytochrome P450 system and it has little effect on the metabolism of other drugs broken down by this system.
As a biological stain and as trypanocide.
Tropisetron was originally developed by Novartis, and rights to the drug were subsequently acquired by Asta Medica (now, part of Meda). In December 1997, Novartis and Kyowa Hakko signed an agreement, pursuant to which the companies would copromote the product in Japan. Tropisetron is currently distributed in various countries worldwide, including Belgium, Germany, Italy, Japan, The Netherlands, Sweden and the U.K.
.
- Muller, W.; T. Stratz (2004). “Local treatment of tendinopathies and myofascial pain syndromes with the 5-HT3 receptor antagonist tropisetron”. Scand J Rheumatic Suppl. 119 (119): 44–48.PMID 15515413. Retrieved 2007-05-17.
- Macor JE, Gurley D, Lanthorn T, Loch J, Mack RA, Mullen G, Tran O, Wright N, Gordon JC (February 2001). “The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist”. Bioorganic & Medicinal Chemistry Letters11 (3): 319–21. doi:10.1016/S0960-894X(00)00670-3.PMID 11212100.
- Cui R, Suemaru K, Li B, Kohnomi S, Araki H (May 2009). “Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: role of alpha7 nicotinic receptors”.European Journal of Pharmacology 609 (1–3): 74–7.doi:10.1016/j.ejphar.2008.12.051. PMID 19374878.
Tropisetron hydrochloride (CAS 105826-92-4)

Application: A potent SR-3 antagonist CAS Number: 105826-92-4 Purity: ≥99% Molecular Weight: 320.82 Molecular Formula: C17H20N2O2 HCl - Navoban™ IS THE FORMULATION
Tropisetron hydrochloride is a potent SR (SR-3) antagonist. It is also a selective, partial agonist at AChR α 7 (α7 nicotinic receptors). Tropisetron hydrochloride is an inhibitor of HTR3.

1alphaH,5alphaH-Tropan-3alpha-yl Indole-3-carboxylate, could be produced through many synthetic methods.
Following is one of the synthesis routes: Indole 3-carbonyl chloride (I) is condensed with endo-8-methyl-8-azabicyclo[3.2.1]octan-3-ol (II) in the presence of butyllithium in THF, or Na2CO3 in the same solvent to produce the final product of Tropisetron.
IMPORTANT REFERENCES
Drugs Fut 1986, 11(2): 106
US 4789673
CN 102532128
CN 102887893
WO 2013123426
WO 2007099069
WO 2009033305
WO 2003032897
WO 2004054552
WO 2005105089
WO 2000048597
WO 2000048581
| CN101033225A | 2 Apr 2007 | 12 Sep 2007 | 北京成宇化工有限公司 | Process of preparing troipisetron |
| CN101787021A | 5 Mar 2010 | 28 Jul 2010 | 王明 | High-purified tropisetron hydrochloride compound |
| US4789673 | 10 Nov 1987 | 6 Dec 1988 | Peter Donatsch | Heterocyclic carboxylic acid amides and esters |
…………………………………………………………………………………………………………
5-HT3 receptor antagonists are a class of compounds which block 5-HT3receptors, and are also sometimes classified as serotonin M receptor antagonists. The 5-HT3 receptor antagonists comprise a defined and recognised class of pharmaceutically active compounds well known in the art and characterised, as their name implies, by their pharmacological activity. Various 5-HT3 receptor antagonist compounds are commercially available and clinically applied, e.g. in the treatment of emesis.
5-HT3 receptor antagonists from various sources have been published for a wide variety of uses, for example for the treatment of visceral pain, migraine, vascular and cluster headache, trigeminal neuralgia, arrhythmia, serotonin-induced gastro-intestinal disorders, including emesis induced by anti-cancer agents, anxiety, stress-related psychiatric disorders, depression, cognitive disorders, social withdrawal, panic attacks, agoraphobia, lung embolism, rhinitis or serotonin-induced nasal disorders, fibromyalgia and local treatment of pain caused by various non-inflammatory or inflammatory conditions. Some have been commercially introduced for the treatment of emesis.
In accordance with the present invention it has now surprisingly been found that 5-HT3 receptor antagonists are useful for the treatment of diseases caused or influenced by activation of thrombocytes.
Thrombocytes play a central role in blood coagulation (clotting) and are therefore also of high importance in the pathogenesis of cardiac infarction and stroke, furthermore also in thrombosis of the veins and inflammatory conditions in the development of atherosclerosis.
Activation of thrombocytes causing blood clotting is based on several mechanisms. It has ■ now been demonstrated that 5-HT3 receptors are present on platelets and that the number of these receptors at the platelet surface is increasing in a dose dependent fashion on addition of ADP (adenosine diphosphate) or TRAP (thrombin receptor activating peptide) known to stimulate thrombocyte activation. The increase of 5-HT3 receptors on addition of compounds inducing aggregation such as ADP and TRAP is proof that such receptors play a role in thrombotic processes. Platelet activity is also important in inflammatory processes in atherosclerotic conditions. Thrombocytes activated by thrombin may, as is already known, induce the production of I L- 1 β , IL-8, MCP (monocyte chemoattractant protein) and other inflammation mediators These are impeded by 5-HT3 receptor antagonists. This demonstrates that 5-HT3 receptor antagonists not only influence blood coagulation but also processes playing a role in the development of atherosclerosis.
Hence, the present invention relates to the use of a 5-HT3 receptor antagonist or of a pharmaceutically acceptable salt of such an antagonist for the manufacture of a pharmaceutical composition for the treatment of a disease caused or influenced by activation of thrombocytes, in particular myocardial infarction, stroke, thrombosis and atherosclerosis.
Any 5-HT3 receptor antagonist can be used in accordance with the invention. Preferred 5-HT3 receptor antagonists which may be employed in accordance with the present invention are ondansetron, 1 ,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1 H-imidazol-1-yl)- methyl]-4H-carbazol-4-one (cf. Merck Index, twelfth edition, item 6979), granisetron, ‘ endo-1-methyl-N-(9-methyl-9-aza-bicyclo[3.3.1]non-3-yl)-1 H-innidazole-3-carboxamide . (cf. loc. cit, item 4557), or dolasetron, 1 H-indole-3-carboxylic acid (2α,6α,8α,9αβ)- octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl ester, (cf. loc. cit., item 3471). ■•
Particular 5-HT3 receptor antagonists which may be employed in accordance with the ■ present invention are those of the formula 1 as defined in European Patent Publication EP O 189 002 B1, in particular tropisetron, indol-S-yl-carboxylic acid-endo-δ-methyl-δ-aza- bicyclo[3,2,1]-oct-3-yl-ester, (cf. loc. cit., item 9914), ramosetron, 4,5,6,7-tetrahydro-5- [(1-methyl-indol-3-yl)carbonyl]benzimidazole (U.S. Pat. No. 5,344,927), fabesetron, (+)- 10-methyl-7-(5-methyl-1 H-imidazol-4-ylmethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-6-one (EP 0 361 317), lintopride, N-(1-ethyl-2-imidazolin-2-y-methyl)-2-methoxy-4-amino-5- chlorobenzamide (Chem. Abstr. No. 107429-63-0), alosetron, 2,3,4,5-tetrahydro-5-methyl- 2-[(5-methyl-1 H-imidazol-4-yl)methyl]-1 H-pyrido[4,3-b]indol-1-one (EP 0 306 323), cilansetron, (-)-(R)-5,6,9,10-tetrahydro-10-[(2-methylimidazol-1-yl)methyl]-4H-pyrido- (3,2,1-jk)carbazol-11(8H)-one, palonosetron, 2-(3S)-1-azabicyclo[2.2.2]oct-3-yl-2,3,3a(S), 4,5,6-hexahydro-1 H-benz(de)isoquinolin-1-one, azasetron, N-(1-azabicyclo[2.2.2]octan-8- yl)-6-chloro-4-methyl-3-oxo-1 ,4-benzoxazine-8-carboxamide, and zatosetron, 5-chloro- 2,2-dimethyl-N-(8-methyl-8-azabicyclo[3.2.1]octan-3-yl)-3H-1-benzofuran-7-carboxamide.
5-HT3 receptor antagonists may be employed in accordance with the invention in free or in pharmaceutically acceptable salt form, e.g. as known in the art, for example, in the case of compounds mentioned above in pharmaceutically acceptable acid addition salt form, for example, in the case of ondansetron as the hydrochloride dihydrate, granisetron as the hydrochloride, dolasetron as the mesylate, tropisetron as the monohydrochloride, ramosetron, fabesetron, alosetron and cilansetron as the hydrochlorides, palonosetron as the monohydrochloride, azasetron as the hydrochloride, and zatosetron as the maleate.
References to 5-HT3 receptor antagonists collectively or individually throughout the present specification and claims are accordingly to be understood as embracing both free compounds and such pharmaceutically acceptable salt forms, e.g. as clinically employed, and further also solvates, e.g. hydrates, or specific crystal forms of any of these compounds or salts.
For use in accordance with the present invention tropisetron (especially in the formulation called Navoban™) is most preferred.
tropisetron hydrochloride is a peripheral neurons and central nervous system 5 – hydroxytryptamine 3 (5-ΗΤ3) receptors potent, highly selective competitive antagonist, mainly by selectively blocking peripheral neurons presynaptic 5-ΗΤ3 receptors inhibit vomiting reflex. For the prevention and treatment of child and adult cancer chemotherapy, radiotherapy and post-operative nausea and vomiting caused.
About tropisetron hydrochloride preparation of patent literature, including (for example): US4797406, US 4789673, CN 101033225, CN 101838266, CN 101787021. Tropisetron hydrochloride preparation generally is this: the firstsynthesis of acid chloride intermediates, and then through esterification acidification, refining and other steps, resulting tropisetron hydrochloride.
The preparation method has, in U.S. Patent (US3980668, US4789673, US4797406, US4803199) described with indole-3 – carboxylic acid with oxalyl chloride, treatment with methylene chloride and n-hexane to give 3 – methyl indole chloride, then with tropenol activation in n-butyl lithium produced under tropenol lithium reaction was treated to obtain tropisetron, and the final hydrochloride salt obtained tropisetron hydrochloride
in which the U.S. patent literature US 4797406 and US 4789673 introduces the indole-3 – carboxylic acid with oxalyl chloride to give 3 – indole chloride then with tropenol activation in n-butyl lithium in the reaction system tray alkyl granisetron. This method uses expensive n-butyl lithium and polluting chloride compound, and the multi-step, only about 20% yield.
Chinese patent literature described in CN 101033225 is improved indolesynthesis _3_ chloro acid as raw material of 1,3 – dimethyl-2 – chloro-imidazoline as the condensing agent, an organic base directly under conditions esterified with tropenol obtain the target product. Although this method avoids the acylation step, a condensing agent in preparing dimethyl-2-chloro-1,3 _ – chloro-imidazoline must use highly toxic phosgene, so there is an obvious lack of this route.
In the literature [tropisetron hydrochloride synthesis improvements, Huaihai Institute of Technology, 2003; 12 (4): 4 wide 43; tropisetron hydrochloride Improved synthesis of Hunan Normal University (Medical Sciences), 2006; 3 (4): 29 ^ 30], respectively, 50%, 52.8% obtained in a yield tropisetron hydrochloride, but still use of expensive n-butyl lithium as a condensation reaction activator.
………………………………………………………………………………………………………………
Example 2:
[0071] The indole-3 – carboxylic acid (20 g, 0. 124mol), benzenesulfonic acid (0. 791g, 0. 005mol), ethyl acetate (230ml), freshly activated 4A molecular sieves (0. 5-1 . Omm) (3 g) was added to a equipped with a thermometer, reflux condenser, 500ml three reaction flask, stir began to heat up, the temperature controlled at 75 ° C _77 ° C, and then began a slow drip Gato Decanter (19. 3 g, 0. 137mol), after the addition was complete the reaction refluxed for 11 hours.
[0072] The reaction was stopped, the organic layer was washed with IOOml a lmol / L of hydrochloric acid, extracting the product three times, the combined aqueous phase was then washed once with 50ml of ethyl acetate. Aqueous phase was 4mol / L sodium hydroxide aqueous solution was adjusted to pH = 9_10, precipitated yellow solid was suction filtered cake was washed with distilled water to neutral, dried under reduced pressure to obtain 33.1 Hector alkoxy granisetron crude. The crude product was dissolved at 60 ° C in 180ml of anhydrous ethanol was slowly added dropwise 12mol / L hydrochloric acid until the pH value 1_2, _5 ° C under crystallization 6 hours, filtered, the filter cake washed with ethanol to white . Drying get tropisetron hydrochloride concept .3 g, yield 71.1% (to indole-3 – carboxylic acid basis), as measured by liquid chromatography purity of 99.56%.
…………………………………………………………………………..
[0016] (3) tropisetron hydrochloride Preparation:
A solution of indole-3 – carbonyl chloride in THF was slowly added dropwise to sodium tropine THF solution, 35 ° C the reaction was stirred overnight, vacuum distillation recovery THF, and recrystallized from 95% ethanol to give a pale yellow solid adding 70mL ethanol, dissolved by heating, cooling pass into the HCl gas at room temperature, the reaction was stirred for 30min, filtered, tropisetron hydrochloride was crude. Recrystallization from absolute ethanol to give white crystalline product 21. 2g (Liquid chromatographic purity 99.91%), total yield of 53.29%, a melting point of view 3185, the product spectrum consistent with the literature.
…………………………………………………………………
Example 5 tropisetron hydrochloride refined
[0063] (a) The IOOg tropisetron hydrochloride was dissolved in 500ml water, then slowly adding 12% (g / ml) of sodium carbonate solution was stirred until the solution PH 8, resulting tropisetron sedimentation, filtration , 40 ° C under reduced pressure and dried to give tropisetron 84. lg.
[0064] (2) obtained in the above Step Tropisetron 84. Ig dissolved in 400ml ethanol, 5. 2g of activated charcoal, stirred at room temperature for 40 minutes, filtered decarbonization, collecting the filtrate.
[0065] (3) The use of the filtrate obtained in step purified by preparative chromatography separation tropisetron hydrochloride refined products, including the column using a mobile phase as the mobile phase volume accounted for 32% of total current volume of methylene chloride and with 68% aqueous hydrochloric acid to pH 1; stationary phase filler is silica; flow rate of 6. 2ml/min; column temperature: 25 ° C. The filtrate was collected, dried under reduced pressure, to obtain purified tropisetronhydrochloride 91. 7g, yield 91. 7%, HPLC purity of 99.9% method.
[0066] Elemental analysis = C17H21ClN2A
[0067] Theoretical value (%): C: 63. 65, H: 6. 60, N: 8. 73, Cl: 11. 05;
[0068] Found (%): C: 63. 62, H: 6. 64, N: 8. 74, Cl: 11. 06.
[0069] UV (MeOH) Amax: 214 (ε 38,222), 229 (ε 17,438), 282 (ε 13,405).
[0070] IR (KCl) CnT1: 3219,2496 (NH), 3103,748 (Ar CH), 2966,1428 (CH), 1692 (C = 0), 1580,1525 (Ar C = C), 1311, 1128 (CN), 1033 (CO).
[0071] 1HNMR (DMSO-D6) δ: 2. 10 (d, 2H, CH2), 2 · 32 (d, 4H, CH2), 2 · 52 (m, 2H, CH2), 2. 68 (s, 3H, CH3), 3. 88 (s, 2H,-CH), 5. 14 (s, H, CH), 8. 03 (m, H, CH), 8. 08 (d, H, CH), 10. 81 (s, H, HCl), 12. 10 (s, H, NH).
[0072] ESI-MS; EI m / z: 285 (M + H, 100%), 284 (M +, 21. 72%) “
…………………………………………………………………………………………………………..
Example 1
[0048] equipped with stirrer, thermometer, condenser IOOOml reaction flask, add 80 grams of tropisetron hydrochloride (I Ci refined products) and 560ml of acetone-water (8:2) mixture, start stirring, heating heated to 60 ° C _65 ° C, until all dissolved clear, incubated for 30 minutes, filtered while hot. The filtrate was cooled to room temperature, and then incubated for 2 hours, crystalline precipitation, filtration, drying ie high purity tropisetron hydrochloride crystals, mp: 280.10C -281.1 ° C, purity 99.96% (HPLC normalization method), the solvent residue testing to meet the requirements.
[0049] Elementary analysis:
[0050] Found (calculated value), C: 63.64 (63.57), H: 6.60 (6.62), N: 8.73 (8.72),
[0051] Cl: 11.05 (11.01);
[0052] X-ray diffraction of the crystal shown in Figure 1. Instrument model and measurement conditions: Rigaku D / max 2500 type diffractometer; CuKa 40Kv 100mA; 2 Θ scan range: 0 – 50 °;
[0053] The infrared spectrum of this crystal is shown in Figure 2, as measured by KBr tablet.
.........................................................................................
7 GALDANSETRON




...............................................................................................................................................................................
7 GALDANSETRON
cas no 116684-92-5
Molecular Formula: C18H19N3O Molecular Weight: 293.36296
GR-81225X
GR-82115C (hydrochloride)
GR-82115C (hydrochloride)
GSK
PRECLINICAL
Nausea and Vomiting, Treatment of
| CA 2081709 EP 0542364 US 4859662 |
GALDANSETRON HYDROCHLORIDE
CAS NO 156712-35-5 (HCl)
Molecular Formula: C18H20ClN3O Molecular Weight: 329.8239
- Galdansetron HCl
- Galdansetron hydrochloride
- GR 81225C
- GR 81225X [as the base]
- UNII-E3M2R8Q947
GALDANSETRON RACEMIC
........................................................................
Patents
- US 20050209293 A
- US 20060024365
- US 4859662 A
- DE 3740352 A1
- WO 2001095902 A1
- WO 2009146537 A1
- US 20100226943 A1
Example 1
(E) -1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methylene]-4H-carbazol-4-one maleate
A solution of 1,2,3,9-tetrahydro-3-[hydroxy [5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl] methyl]-9-methyl-4H-carbazol-4-one (2.70 g) in glacial acetic acid (100 ml) was treated with p-toluenesulfonic acid monohydrate (10.80 g) and the stirred solution was heated to reflux for 4 hours. The cool dark liquid was evaporated, treated with aqueous saturated sodium bicarbonate solution (250 ml) and extracted into ethyl acetate (4 × 250 ml). The combined, dried organic extracts were evaporated and purified by SPCC. The eluting with System A (978: 20: 2 → 945: 50: 5) afforded the free base of the title compound as a light yellow-brown solid (488 mg). A hot solution of the free base (87 mg) in ethanol, about 16 ml) was treated with a hot solution of maleic acid (38 mg) in ethanol (1 ml). After cooling, the precipitate was collected to give the title compound (81 mg), mp 205-209 ° was obtained.
Analysis found: C: 65.1, H 5.2, N 10.2; C ₁ ₈ H ₁ ₇ N ₃ O · C ₄ H ₄ O ₄
theoretical values: C: 64.9, H 5.2, N 10.3%.
theoretical values: C: 64.9, H 5.2, N 10.3%.
Example 7 1,2,3,9-tetrahydro-9-methyl-3-[(1H-imidazol-4-yl) methylene] - 4H-carbazol-4-one
A solution of diisopropylamine (1.54 ml) in dry THF (20 ml) of -78 ° was treated dropwise with n-butyllithium (1.32 M in hexane, 8.3 ml). The mixture was allowed to warm to 0 ° and cooled to -78 ° again. It was then in the course of 3 minutes to a stirred suspension of 1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one (2.0 g) in dry THF (80 ml) of -78 optionally °. The resulting suspension was stirred at -78 ° on this for 2 hours and then treated with 1 - treated (triphenylmethyl)-1H-imida zol-4-carboxaldehyde (3.72 g). The mixture was stirred for a further 2 hours, during which time it was allowed to warm slowly to room temperature. Then it was cooled to -78 ° and quenched with acetic acid (2 ml). The resulting solution was allowed to warm to room temperature and 8% aqueous sodium bicarbonate solution (600 ml) was poured.
The mixture was extracted with dichloromethane (3 x 150 ml) and the combined, dried organic extracts were evaporated to give a foam. A solution of this foam and p-toluenesulfonic acid monohydrate (18 g) in a mixture of glacial acetic acid (25 ml) and dry THF (150 ml) was heated for 5 hours under reflux. The cooled mixture was carefully added to an 8% aqueous sodium bicarbonate solution (650 ml) and extracted with dichloromethane (3 x 150 ml). The combined, dried organic extracts were evaporated to give a solid which was obtained by FCC eluting with System A (100: 1: 10) was purified. In this way the title compound (1.42 g), mp 225-232 ° was obtained.
Analysis found: C: 73.3, H 5.6, N 14.7; C ₁ ₇ H ₁ ₅ N ₃ O
theoretical values: C: 73.6, H 5.5, N 15.1%
theoretical values: C: 73.6, H 5.5, N 15.1%
Example 8
1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one maleate
A solution of 1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methylene]-4H-carbazol-4-one (3.50 g) in DMF (85 ml) and ethanol (50 ml) was added to a prereduced suspension of 10% palladium on carbon added (3.4 g) in ethanol (50 ml) and hydrogenated at room temperature and atmospheric pressure until hydrogen uptake ceased (270 ml). The catalyst was filtered off and the filtrate was evaporated. The residue was adsorbed from methanol (170 ml) of SPCC-FCC silica and applied to a column. A gradient elution system A (967: 30: 3 → 912: 80: 8) afforded the free base of the title compound as a solid (2.32 g). A portion of this solid (500 mg) in hot ethanol (15 ml) was treated with a hot solution of maleic acid (224 mg) in ethanol (2 ml). Upon cooling, a precipitate was collected, the title compound (415 mg), mp 130.5-137 ° revealed. tlc (system A 200: 10: 1) 0.30.
Analysis found: C: 63.2, H 5.5, N 9.7; C ₁ ₈ H ₁ ₉ N ₃ O · C ₄ H ₄ O ₄ · 0.33 H ₂ O
theoretical values: C: 63.6, H 5.7, N 10.1%. Water sample found: 1.55% wt. / Wt. 0.33 mol H ₂ O ≡
theoretical values: C: 63.6, H 5.7, N 10.1%. Water sample found: 1.55% wt. / Wt. 0.33 mol H ₂ O ≡
¹ H-NMR (d ⁶-DMSO) δ 1.8-1.98 (1H, m), 2.1-2.25 (1H, m), 2.25 (3H, s), 2.68 to 2, 84 (2H, m), 2.85 to 3.3 (3H, m), 3.75 (3H, s), 6.0 (2H, s-maleate), 7.18-7.32 (2H, m), 7.57 (1H, brd), 8.03 (1H, brd), 8.88 (1H, s).
+FORM
Example 31
(+) -1,2,3,9-Tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one
A solution of (±) -1,2,3,9-tetrahydro-9-methyl-3-[(5-me thyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one (500 mg) in warm methanol (30 ml) was treated with a solution of (+) -2,3-bis [[(4-methylphenyl) carbonyl] oxy] butanedioic acid (690 mg) in methanol (10 ml), and The solution was allowed to stand for 3 days at 0 °. It was then filtered to give a solid remained which was recrystallized from methanol to give the desired salt (195 mg), mp 146-148 ° was obtained. A part of this salt (186 mg) was suspended (10 ml) in water and treated with potassium carbonate (79.2 mg) and the mixture was extracted with dichloromethane (2 x 40 ml). The combined, dried organic extracts were evaporated in vacuo to give the title compound (79.2 mg) as a solid, mp 230-232 ° stayed behind. [Α] = 49.7 ° (c = 0.41%, CHCl ₃).
- FORM
Example 32
(-) -1,2,3,9-Tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one
A solution of (±) -1,2,3,9-tetrahydro-9-methyl-3-[(5-me thyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one (500 mg) in warm methanol (30 ml) was added a solution of (- treated) -2,3-bis [[(4-methylphenyl) carbonyl] oxy] butanedioic acid (690 mg) in methanol (10 ml), and The solution was allowed to stand at 0 ° for 3 days. It was filtered, producing a solid remained which was recrystallized from methanol to give the desired salt (162 mg), mp 147-148 ° was obtained. This was suspended in water (15 ml) and treated with a solution of potassium carbonate (1 g in 10 ml water). The mixture was extracted with dichloromethane (2 x 30 ml). The combined, dried organic extracts were evaporated in vacuo to give the title compound (72.5 mg) as a solid, mp 230-232 ° stayed behind. [Α] = 48.4 ° (c = 0.44%, CHCl ₃).
Example 33
1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) - methyl]-4H-carbazol-4-one
A solution of Intermediate 7 (190 mg) in dry DMF (1 ml) was added dropwise to a stirred suspension of sodium hydride, under nitrogen (52% dispersion in oil, 20 mg) in dry DMF (0.4 ml). After 15 minutes, iodomethane (0.027 ml) was added and the mixture was stirred for 1.5 hours. Water (20 ml) was added and the suspension was extracted with dichloromethane (3 x 10 ml). The combined, dried organic extracts were evaporated to give an oil (ca. 300 mg) in a mixture of THF (4 ml), acetic acid (4 ml) and water (4 ml) was dissolved. The mixture was heated at reflux for 1.5 hours. It was poured (20 ml) in saturated potassium carbonate solution and extracted with dichloromethane (3 x 10 ml). The combined, dried organic extracts were evaporated to give a semi-solid (ca 255 mg) was obtained by SPCC eluting with System A (200: 1: 10) was purified. In this way the title compound (7 mg) was obtained. The ¹ H NMR and tlc of this material were values with the corresponding values of the product of Example 8 in line.
Example 34
1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) - methyl]-4H-carbazol-4-one
n-Butyllithium (1.45M in hexane; 2.07 ml) was added dropwise to a cold (-70 (20 ml) under nitrogen. The solution was allowed to reach 0 30 min, cooled to -70 solution of) 1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one (500 mg) in dry THF (10 ml under nitrogen. Hexamethylphosphoramide (0.44 ml) was added and the mixture was allowed to reach 0 cooled to -70 4-(chloromethyl)-5-methyl-1-(triphenylmethyl) -1H-imidazole (936 mg) in dry THF (15 ml) was added and the mixture was allowed to reach ca. 20 sodium bicarbonate solution (100 ml) and extracted with dichloromethane (3 give a semi-solid which was treated with a mixture of acetic acid (10 ml), water (10 ml) and THF (10 ml) and heated at reflux for 1.5 h. The solution was poured into saturated potassium carbonate solution (100 mml) and extracted with dichloromethane (3 organic extracts were evaporated to give a solid (ca. 1.8 g) which was purified by SPCC eluting with System A (200:10:1) to give the title compound (17 mg). The .sup.1 H-n.m.r. and t.l.c. of this material were consistent with those obtained from the product of Example 8.
BREAKING OF MALEATE SALT
Example 35
1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one
1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4-yl) - methyl]-4H-carbazol-maleate (37 mg) was partitioned between 2 N sodium bicarbonate solution (10 ml) and chloroform (3 x 15 ml) separated. The combined, dried organic layers were evaporated to give the free base (26 mg) was obtained, which was dissolved at -10 ° under nitrogen in 10% aqueous THF (4 ml). To this stirred solution, a solution of 2,3-dichloro-5 ,6-dicyano-1 ,4-benzoquinone (49 mg) was added in dry THF (1.6 ml) was added dropwise, and the reaction mixture was added over 3 hours allowed to warm to 0 °. The solution was evaporated in vacuo and purified by FCC eluting with System A (94.5: 5: 0.5) to give the title compound (10 mg) was obtained as a solid.The ¹ H NMR and tlc values of this material were consistent with the corresponding values of the product of Example 8
INTERMEDIATE 7
Intermediate 7
1,2,3,9-Tetrahydro-3-[(5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl) methyl]-4H-carbazol-4-one
A solution of triphenylchloromethane (4.2 g) in dry DMF (40 ml) was added dropwise to a solution of 1,2,3,9 - tetrahydro-3-[(5-methyl-1H-imidazol-4-yl) methyl ]-4H-carbazol-4-one (3.5 g) and triethylamine (1.75 ml) in dry DMF (35 ml) under nitrogen.After stirring for 4 hours the mixture was poured (300 ml) in water and extracted with dichloromethane (3 x 100 ml). The combined extracts were washed with water (200 ml), dried and evaporated to give an oil (about 9 g) was obtained. This was purified by FCC eluting with System A (200: 1: 10) to give the title compound (4.57 g) as a foam, tlc (system A 200: 10: 1), Rf 0.32 was obtained.
Intermediate 8
4 - (chloromethyl)-5-methyl-1-(triphenylmethyl)-1H-imidazole
A solution of thionyl chloride (1.3 ml) in dry dichloromethane (10 ml) was added over 5 minutes to a stirred suspension of 5-methyl-1-(triphenylmethyl) - 1H-imidazol-4-methanol (5.0 g ) in a mixture of dichloromethane (100 ml) and dry DMF (2 ml) at 0 °. The mixture was stirred at 0 ° for 30 minutes and washed successively with 8% sodium bicarbonate (2 x 50 ml), water (50 ml), dried and evaporated in vacuo below 40 ° to give an oil (5 g) was obtained. This was dissolved in ether (100 ml), and the resulting solution was filtered through a silica pad which was eluted with ether (2 x 100 ml) further. The combined filtrates were evaporated below 40 °, whereby a foam was obtained which was triturated with cold hexane and filtered. There was thus obtained the title compound (4.2 g) as a solid, mp 133-135 °, was obtained
Intermediate 14
1,2,3,9-tetrahydro-9-methyl-3 [[5-methyl-1-(triphenylmethyl) - 1H-imidazol-4-yl] methyl]-4H-carbazol-4-one
A solution of triphenylchloromethane (286 mg) in dry DMF (10 ml) was added dropwise to a stirred solution of 1,2,3,9-tetrahydro-9-methyl-3-[(5-methyl-1H-imidazol-4 - optionally yl) methyl]-4H-carbazol-4-one (292 mg) and triethylamine (101 mg) in dry DMF (20 ml). The resulting solution was stirred for 3.5 hours at room temperature under nitrogen. The reaction mixture was then poured into water (100 ml) and the resulting suspension was extracted with dichloromethane (3 x 50 ml). The combined, dried organic extracts were adsorbed onto FCC silica, which was then applied to a column. FCC eluting with System A (150: 8: 1) afforded a solid which, by crystallization from dichloromethane: hexane (2: 1) was further purified to give the title compound (304 mg), mp 193 to 195 °, was obtained.
INTERMEDIATES
CAS 27387-31-1
- C13 H13 N O
- 4H-Carbazol-4-one, 1,2,3,9-tetrahydro-9-methyl-
- Carbazol-4(1H)-one, 2,3-dihydro-9-methyl-
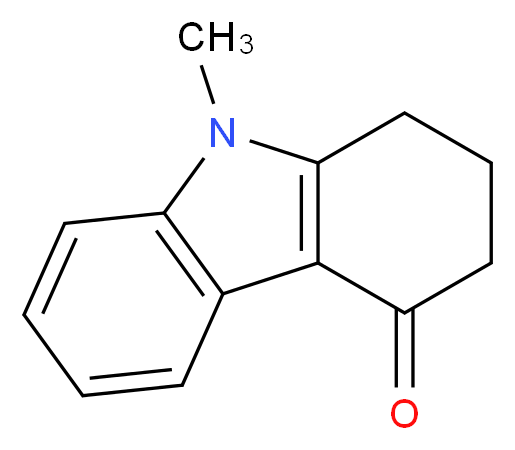
- INTERMEDIATE 2
- CAS 113140-81-1
-
- C24 H20 N2 O
- 1H-Imidazole-4-carboxaldehyde, 5-methyl-1-(triphenylmethyl)
- INTERMEDIATE 3
- CAS : 116684-96-9
- C37 H33 N3 O2
- 4H-Carbazol-4-one, 1,2,3,9-tetrahydro-3-[hydroxy[5-methyl-1-(triphenymethyl)-1H-imidazol-4-yl]methyl]-9-methyl-
- 4H-Carbazol-4-one, 1,2,3,9-tetrahydro-3-[hydroxy[5-methyl-1-(triphenylmethyl)-1H-imidazol- 4-yl]methyl]-9-methyl-
- INTEMEDIATE 4
- triphenylchloromethane
- CAS 76-83-5
- INTERMEDIATE 5
- 4-(chloromethyl)-5-methyl-1-(triphenylmethyl) -1H-imidazole
- ....................................................................................................
8 AZASETRON
AZASETRON, NAZASETRON
N-(1-azabicyclo[2.2.2]octan-8-yl)-6-chloro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide
6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide
123039-99-6 , 123040-69-7 (free base)
141922-90-9 HYD SALT
Y-25130 (hydrochloride)LAUNCHED 1994 AS HCL SALT FORM SEROTONE
a selective 5-HT3 receptor antagonist , ANTIEMETIC
- UNII-77HC7URR9Z
Mitsubishi Tanabe Pharma, JAPAN TOBACCO (Originator)
FOR..Nausea and Vomiting, Treatment of
Prokinetic Agents
Prokinetic Agents

Chemical and Pharmaceutical Bulletin, 1992 , vol. 40, 3 p. 624 – 630, ENTRY 15 , MP 305 OF HCL SALT
Biological and Pharmaceutical Bulletin, 2006 , vol. 29, 9 p. 1931 – 1935, AS MERCK 903
US 4892872…
CN 101786963…
WO 1996001630..
WO 2006006595..
WO 2007134077..
CN 102526740, CN 102451166, US 4892872, JP 2008297277
US5773436 A1, WO2006/119295 A2, EP1336602 A1, US4892872 A1,
US2003/18008 A1, US2002/147197 A1, US4892872
 AZASETRON
AZASETRON

The nitration of 5-chloro-2-hydroxybenzoic acid methyl ester (I) with nitric acid in sulfuric acid gives 5-chloro-2-hydroxy-3-nitrobenzoic acid methyl ester (II), which is reduced with Fe and NH4Cl in water yielding 3-amino-5-chloro-2-hydroxybenzoic acid methyl ester (III). The cyclization of (III) with chloroacetyl chloride (IV) by means of NaHCO3 in CHCl3 – water affords 6-chloro-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-8-carboxylic acid methyl ester (V), which is methylated with methyl iodide and K2CO3 in DMF affording the corresponding 4-methyl derivative (VI). Hydrolysis of (VI) with ethanolic NaOH gives the corresponding acid (VII), which by treatment with refluxing SOCl2 is converted into its acyl chloride (VIII). Finally, this compound is condensed with 3-aminoquinuclidine (IX) by means of N-methylmorpholine (NMM) in CHCl3.
………………
SYNTHESIS
Azasetron hydrochloride (Azasetron hydrochloride) is a secondary cancer drugs, mainly synthetic route is as follows
[0003]

[0004] In EP 0313393; JP 1989207290; JP 1990005415; US 4892872; Eur.Pat.Appl., 313393,, Chem pharm Bull, 1992,40 (3) :624-630 reported the synthesis of intermediates III is with ammonium chloride, iron as the reducing agent, the nitro substrate was dissolved in toluene and added dropwise to a reduction of the media inside, the study found that this approach has a number of disadvantages, notably the reaction can not be completely, while post-processing is very troublesome, purity of the product obtained is poor, and difficult purification. Although the “Chinese Medicinal Chemistry” magazine, 10 (2) ,138-140; 2000, the author hydrochloride instead of ammonium chloride, but still need the nitro group was dissolved in toluene was added dropwise, in fact, the nitro compound in toluene the solubility is limited, and in the process will precipitate dropwise addition, there are also incomplete reaction, impurities, and complicated post-treatment problems.
[0005] In the “China Pharmaceutical Industry” magazine 34 (3) ,214-215; 2003 in order to restore hydrosulfite as reductant,
Azasetron preparation of intermediates, the steps are as follows:
[0015] (I) A mixture of 3 – nitro-5 – chloro-salicylate added glacial acetic acid was stirred for 30-40 minutes, add water, temperature 30 ~ 100 ° C, adding iron powder, 2.5-3 hour plus finished, the addition was completed, at a temperature of 30 ~ 100 ° C the reaction for 5 to 10 hours; give 3 – amino-5-chloro-salicylate mixture;
[0016] The mass ratio of the added material is 3 – nitro-5 – chloro-salicylate: acetic acid: water: iron = 1: 5 ~ 10: 5 ~ 10: 0.5 ~ I;
[0017] (2) obtained in the step ⑴ added to a mixture of glacial acetic acid, glacial acetic acid added in the amount of step ⑴ same amount of glacial acetic acid, 70 ° C was stirred for 30-35 minutes, filtered through celite to remove the iron sludge to obtain filter cake; this procedure to that step (I) the resulting product was dissolved in glacial acetic acid solvent sufficient, in order to facilitate post-processing; while removing impurities.
[0018] (3) The filter cake was washed with an organic solvent, the combined filtrate and stirred for 10-15 minutes, allowed to stand for 30-35 minutes, the organic layer was separated; aqueous phase was extracted once again with an organic solvent, the combined organic solution was washed three times , mention made three – amino -5 chloro methyl salicylate (intermediate III) sulphate solution.
[0019] (4) of step (3) of 3 – amino-5-chloro-salicylate was added a saturated NaHCO3 solution, saturated NaHCO3 solution and the amount of acetic acid in step ⑴ same volume, cooling to _1 ° C ~ 0 ° C, control the temperature -1 ° C_5 ° C was added dropwise acetyl chloride, drop Bi. Reaction 2-3 hours. To give 3 – (2 – chloro-acetylamino)-5-chloro-mixed solution of methyl salicylate;
[0020] The amount of chloroacetyl chloride is added in step ⑴ source material 3 – nitro-5 – chloro molar ratio of methyl salicylate
I: 0.9-0.95.
[0021] (5) obtained in the step ⑷ 3 – (2 – chloro-acetylamino) methyl salicylate _5 chloride mixture was heated to 30-40 ° C, stirred for 1-1.5 hours; organic layer was separated, water phase extracted once again with an organic solvent, the combined organic layer was washed with water three times, separated and the organic layer is directly subjected to atmospheric distillation, the organic solvent is distilled off, distillation was complete, methanol or ethanol as the crystallization solvent, 80 ° C under stirring for 2.5 hours under reflux , to give 3 – (2 – chloro-acetylamino) _5 chlorine in alcohol solution of methyl salicylate. This step is intended to 3 – (2 – chloro-acetylamino)-5-chloro methyl salicylate purification.
[0022] (6) in the step (5) cooling to room temperature, an alcoholic solution, crystallization, centrifugation, and washed with ethanol crystal; 80 ° C drying in needle-like crystals 3 – (2 – chloro-acetylamino) _5 Chlorine methyl salicylate (intermediate IV).
……………………………………………………
USEFUL PATENTS
6-31-1998
|
Use of serotonin antagonists for treating fibromyalgia
| |
11-28-1997
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
| |
11-28-1997
|
DHA-PHARMACEUTICAL AGENT CONJUGATES DHA-PHARMACEUTICAL AGENT CONJUGATES
| |
11-28-1997
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
| |
3-28-1997
|
5-HT3 RECEPTOR ANTAGONISTS FOR DYSKINESIA
| |
2-19-1997
|
Nasally administrable compositions
| |
1-26-1996
|
PREVENTIVE OR REMEDY FOR IRRITABLE BOWEL SYNDROME OR DIARRHEA
| |
3-17-1994
|
Benzoxazine compounds and pharmaceutical use thereof.
| |
1-10-1990
|
Benzoxazine compounds and pharmaceutical use thereof
|
12-17-1999
|
MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT>3< RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS
| |
11-17-1999
|
Use of serotonin antagonists for treating fibromyalgia
| |
11-5-1999
|
CNRE BINDING FACTORS AND USES THEREOF
| |
7-23-1999
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
| |
7-7-1999
|
Taxane compounds and compositions
| |
6-25-1999
|
ORAL DELIVERY FORMULATION
| |
4-16-1999
|
MEDICAMENTS MEDICAMENTS
| |
2-5-1999
|
CHEMOTHERAPY SYNERGISTIC AGENT
| |
10-30-1998
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
| |
8-19-1998
|
DHA-pharmaceutical agent conjugates of taxanes
|
8-7-2009
|
ALISKIREN MODULATION OF NEUROGENESIS
| |
7-3-2003
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
| |
9-18-2002
|
Inhibition of emetic effect of metformin with 5-HT3 receptor antagonists
| |
11-8-2001
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
| |
6-14-2001
|
Nasally administrable compositions
| |
12-22-2000
|
RECEPTOR AGONISTS AND ANTAGONISTS COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION
| |
12-15-2000
|
PHARMACEUTICAL COMPOSITION FOR INTRANASAL USE OF ACTIVE SUBSTANCES THAT ARE INSOLUBLE AND/OR HARDLY SOLUBLE IN WATER
| |
11-31-2000
|
ANTI-TUMOR COMPRISING BOROPROLINE COMPOUNDS
| |
11-30-2000
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
|
11-17-2000
|
FATTY ACID-N-SUBSTITUTED INDOL-3-GLYOXYL-AMIDE COMPOSITIONS AND USES THEREOF
| |
10-12-2000
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
| |
9-15-2000
|
FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF
| |
7-20-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS
| |
7-7-2000
|
USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE
| |
6-28-2000
|
Taxanes
| |
5-32-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS
| |
1-14-2000
|
NOVEL INDOLOCARBAZOLE DERIVATIVES USEFUL FOR THE TREATMENT OF NEURODEGENERATIVE DISEASES AND CANCER
| |
1-12-2000
|
Indolocarbazole derivatives useful for the treatment of neurodegenerative diseases and cancer
| |
12-30-1999
|
METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS
|
……….. AZASETRON
AZASETRON
AZASETRON
NMR OF HCL SALT
…………….
SYNTHESIS
EXAMPLE 15
To a solution of 3.0 g of 3-aminoquinuclidine and 3.0 g of N-methylmorpholine in 60 ml of chloroform is added 6.2 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-2H-1,4-benzoxazine-8-carboxylic acid chloride under cooling and stirring followed by stirring for 2 hours. The resultant solution is washed with water, aqueous sodium hydrogen carbonate and then water, and dried over magnesium sulfate. After the solvent is distilled off under reduced pressure, the residue is recrystallized from ethanol-isopropyl ether and treated with ethanolic hydrochloric acid to give 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide hydrochloride, melting at 281
………..
SYNTHESIS AND + AND – ISOMERS
EXAMPLE 36
A solution of 6 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide and 2.7 g of D-(-)-tartaric acid in 100 ml of methanol and 200 ml of ethanol is allowed to stand. The precipitated crystals are collected by filtration and recrystallized from methanol repeatedly to give the R-(+) isomer, [α].sub.D.sup.25 =+36.72 (c=1, chloroform), melting at 177 with ethanolic hydrochloric acid, and then the precipitated crystals are collected by filtration and dried to give the R-(+) isomer hydrochloride, [α].sub.D.sup.25 =+1.4 (c=1, water), melting at 309
By using L-(+)-tartaric acid in a similar manner, the R-(-) optical isomer, [α].sub.D.sup.25.5 =-36.76 (c=1, chloroform), melting at 176 [α].sub.D.sup.25.5 =-1.2 (c=1, water), melts at 310
...................................................................................

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
..................................................

back to home for more updates
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
No comments:
Post a Comment